|
|
|
|
|
|
http://www.geneclinics.org/profiles/androgen/details.html
Androgen Resistance Syndrome, Testicular Feminization. Includes: Complete Androgen Insensitivity Syndrome (CAIS), Partial Androgen Insensitivity Syndrome (PAIS), Mild Androgen Insensitivity Syndrome (MAIS)]
|
Authors:
|
Bruce Gottlieb, PhD
Lenore K Beitel, PhD Mark A Trifiro, MD |
Disease characteristics. Androgen insensitivity syndrome (AIS) is typically characterized by evidence of feminization (i.e., undermasculinization) of the external genitalia at birth, abnormal secondary sexual development in puberty, and infertility in individuals with a 46,XY karyotype. AIS represents a spectrum of defects in androgen action and can be subdivided into three broad phenotypes: complete androgen insensitivity syndrome (CAIS), with typical female genitalia; partial androgen insensitivity syndrome (PAIS) with predominantly female, predominantly male, or ambiguous genitalia; and mild androgen insensitivity syndrome (MAIS) with typical male genitalia.
Diagnosis/testing. The diagnosis of AIS in individuals with a 46,XY karyotype is based on the following clinical findings: undermasculinization of the external genitalia, impaired spermatogenesis with otherwise normal testes, absent or rudimentary müllerian structures, evidence of normal or increased synthesis of testosterone and its normal conversion to dihydrotestosterone, normal or increased luteinizing hormone (LH) production by the pituitary gland, and deficient or defective androgen-binding activity of genital skin fibroblasts. Molecular genetic testing of the AR gene, the only gene known to be associated with androgen insensitivity syndrome, detects mutations in more than 95% of probands with complete androgen insensitivity and is available clinically. Its yield in individuals with partial or mild forms of AIS is unknown.
Management. To prevent testicular malignancy, treatment of CAIS includes either removal of the testes after puberty when feminization is complete or prepubertal gonadectomy accompanied by estrogen replacement therapy. Additional treatment for CAIS may include vaginal dilatation to avoid dyspareunia. Treatment of PAIS in individuals with predominantly female genitalia is similar to treatment of CAIS but is more likely to include prepubertal gonadectomy to help avoid increasing clitoromegaly at the time of puberty. In individuals with PAIS and ambiguous or predominantly male genitalia, parents and healthcare professionals should assign sex of rearing as early as possible in infancy. Those individuals with PAIS who are raised as males may undergo urologic surgery such as orchiopexy and hypospadias repair. Those individuals with PAIS who are raised as females and who undergo gonadectomy after puberty may need combined estrogen and androgen replacement therapy. Males with MAIS may require mammoplasty for gynecomastia. A trial of androgen pharmacotherapy may help improve virilization in infancy. Systematic disclosure of the diagnosis of AIS in an empathic environment, with both professional and family support, is encouraged. Surveillance includes periodic reevaluation for gynecomastia during puberty in individuals assigned a male sex.
Genetic counseling. AIS is inherited in an X-linked recessive manner. Affected 46,XY individuals are almost always infertile. Carrier females have a 50% chance of transmitting the AR gene mutation in each pregnancy. Carrier testing is available on a clinical basis once the AR mutation has been identified in an affected family member. Prenatal molecular genetic testing is possible for pregnancies of women who are known carriers of the AR mutation present in the family.
Androgen insensitivity syndrome (AIS) can be subdivided into three phenotypes: complete androgen insensitivity syndrome (CAIS), partial androgen insensitivity syndrome (PAIS), and mild androgen insensitivity syndrome (MAIS) (Table 1).
The clinical findings that permit a presumptive diagnosis of AIS include the following:
|
||||||||||||||||
|
Adapted from
Sinnecker et al 1997
|
The diagnosis of CAIS is usually made on clinical findings and laboratory evaluations alone.
The diagnosis of PAIS and MAIS may also require a family history consistent with X-linked inheritance, as laboratory findings useful in establishing the diagnosis may not be present in all affected individuals [Gottlieb, Pinsky et al 1999].
The laboratory findings required for the diagnosis of AIS include the following:
46,XY karyotype
Evidence of normal or increased synthesis of testosterone (T) by the testes
Evidence of normal conversion of testosterone to dihydrotestosterone (DHT)
Evidence of normal or increased luteinizing hormone (LH) production by the pituitary gland
In CAIS but not in PAIS: possible reduction in postnatal (0-3 months) surge in serum LH and serum T concentrations [Bouvattier et al 2002]
Evidence of deficient or defective androgen-binding activity of genital skin fibroblasts
Family history. The diagnosis of CAIS can be established by clinical and laboratory findings alone; however, the diagnosis of PAIS and MAIS may require a family history of other affected individuals related to each other in a pattern consistent with X-linked recessive inheritance. "Other affected family members" refers to:
Affected 46,XY individuals
Manifesting female (46 XX) carriers. About 10% of carrier females are manifesting carriers with asymmetric distribution and sparse or delayed growth of pubic and/or axillary hair.
Additional findings in affected individuals with no family history of the syndrome that substantiate the apparent diagnosis of PAIS in an individual with the "predominantly male" phenotype (Table 1):
Impaired development of the prostate and of the Wolffian duct derivatives demonstrated by ultrasonography or genitourography
Less-than-normal decline of sex hormone-binding globulin (SHBG) in response to a standard dose of the anabolic androgen, stanozolol [Sinnecker et al 1997]
Higher-than-normal levels of anti-müllerian hormone during the first year of life or after puberty has begun
GeneReviews designates a molecular genetic test as clinically available only if the test is listed in the GeneTests Laboratory Directory by at least one US CLIA-certified laboratory or a clinical laboratory outside the US. GeneTests does not independently verify information provided by laboratories and does not warrant any aspect of a laboratory's work; listing in GeneTests does not imply that laboratories are in compliance with accreditation, licensure, or patent laws. Clinicians must communicate directly with the laboratories to verify information. —ED.
Gene. AR is the only gene known to be associated with androgen insensitivity syndrome.
Molecular genetic testing: Clinical uses
Diagnosis
Molecular genetic testing: Clinical method
Sequence analysis. Sequencing of all eight exons of the AR gene detects mutations in more than 95% of individuals with CAIS. The mutation detection rate for milder phenotypes is not known; however, it is less than 50% for PAIS and even less for MAIS.
In the presence of deficient or defective androgen-binding activity in genital skin fibroblasts, the likelihood of finding a mutation in the androgen-binding domain of the AR gene approaches 40% [Weidemann et al 1996]. In the presence of normal androgen binding in genital skin fibroblasts, the likelihood of finding an AIS-causing mutation in the AR gene is 10% or less, even when exon 1 is screened and/or sequenced in its entirety.
Table 2 summarizes
molecular genetic testing for this syndrome.
|
Test Method
|
Mutations Detected
|
Mutation Detection Rate
|
Test Availability
|
|---|---|---|---|
|
Clinical
|
Interpretation of test results
In individuals with decreased or defective androgen-binding activity, some of the negative mutation searches reflect:
Covert mutations in regulatory or deep intronic portions of the AR gene that are not identifiable with the clinically available tests [Gottlieb, Vasiliou et al 1999]:
A "timing" problem in an individual with a normal AR gene; i.e., the acquisition of normal testosterone synthesis or normal androgen responsiveness is delayed beyond the critical periods for normal external and/or internal male genital differentiation.
Mutations may be present in genes whose products either collaborate with the AR gene or are subject to androgenic control [Cheung-Flynn et al 2005].
Somatic mosaicism of the AR gene mutations could result in mutations being present in DNA in genital skin and not in blood [Gottlieb et al 2001b].
For other issues to consider in interpretation of sequence analysis results, click here .
Perform sequence analysis of the AR gene on DNA extracted from a blood sample from the affected individual. Note: It is helpful if a blood sample from the mother is also available for sequence analysis.
If no AR mutation is found, test a biopsy of genital skin for defective androgen binding.
Expansion of the polymorphic CAG repeat within the AR gene causes spinobulbar muscular atrophy (SBMA; Kennedy disease).
Other disease conditions associated with alterations to the AR gene [Gottlieb, Beitel, Wu, Elhaji et al 2004] include the following:
Prostate cancer
Male breast cancer
Laryngeal cancer
In addition, variations in length of CAG repeat in the AR gene have been associated with the following conditions:
Prostate cancer
Male infertility
Female breast cancer
Endometrial cancer
Colon cancer
Complete androgen insensitivity syndrome (CAIS; testicular feminization; Tfm). Individuals with CAIS have normal female external genitalia. They typically present either before puberty with masses in the inguinal canal that are subsequently identified as testes or at puberty with primary amenorrhea and sparse to absent pubic or axillary hair. Breasts and female adiposity develop normally. Sexual identity and orientation are unaffected.
CAIS almost always runs true in families; that is, affected XY relatives usually have normal female external genitalia and seldom have any sign of external genital masculinization, such as clitoromegaly or posterior labial fusion [Boehmer et al 2001]. On occasion, Wolffian duct development is observed [Hannema et al 2004].
Partial AIS (PAIS) with predominantly female external genitalia (Table 1) presents in a manner similar to CAIS; however, affected individuals have signs of external genital masculinization including clitoromegaly or posterior labial fusion.
Partial AIS with ambiguous genitalia or predominantly male genitalia (PAIS; Reifenstein syndrome). Determining the sex of rearing may be an issue for children with frank genital ambiguity. In families with PAIS, phenotypic disparity may warrant opposite sexes-of-rearing [Rodien et al 1996 , Evans et al 1997 , Boehmer et al 2001]. Individuals with PAIS and predominantly male genitalia are raised as males. Gynecomastia at puberty and impaired spermatogenesis occur in all individuals with PAIS. Pubic hair is usually moderate; facial, body, and axillary hair are often reduced.
Mild AIS (MAIS; undervirilized male syndrome). The external genitalia of affected individuals are unambiguously male. They usually present with gynecomastia at puberty. They may have undermasculinization that includes sparse facial and body hair and small penis. Impotence may be a complaint. Spermatogenesis may or may not be impaired. In some instances, the only observed abnormality appears to be male infertility [Gottlieb et al 2005]; therefore, MAIS could explain some idiopathic male infertility.
MAIS almost always runs true in families.
A correlation does exist among certain missense AR mutations, their functional consequences, and external genital development particularly in the case of CAIS (see the Androgen Receptor Gene Mutations Database).
The correlation is much less clear in PAIS, in which interfamilial phenotypic variation is observed [Brinkmann et al 2000 , Boehmer et al 2001 , Deeb et al 2005].
In addition, over 25 instances in which identical AR mutations produce different AIS phenotypes are listed in the AR database [Gottlieb et al 2001a].
In some instances, the variable expressivity associated with a number of point mutations may be attributed to somatic mosaicism rather than to the modifying influence of "background" genetic factors [Boehmer et al 1997 , Holterhus et al 1997 , Holterhus et al 2001 , Kohler et al 2005]. A detailed discussion of the possible role of somatic mosaicism as a cause of variable expressivity is presented by Gottlieb et al (2001b).
It remains to be determined whether specific missense mutations can be correlated with normal or impaired spermatogenesis and with absence or presence of localized expressions of undervirilization such as gynecomastia, high-pitched voice, and impotence.
In addition to causing different forms of AIS, AR gene mutations have also been associated with cancers and prostate cancer in particular [Gottlieb, Beitel, Wu, Elhaji et al 2004]. The allelic variants associated with cancer, however, appear to result in a gain of function rather than loss of function as seen in AIS.
No definitive data regarding penetrance exist, possibly because of under-ascertainment of affected individuals, particularly phenotypic but infertile males in whom AR molecular genetic testing may not be performed [Gottlieb et al 2005].
No definitive data exist for AIS.
The terms "testicular feminization" and androgen resistance syndrome are outdated and thus rarely used now.
Standard references quote prevalence of 2:100,000 to 5:100,000 for complete AIS (CAIS); based on estimates derived from otherwise healthy phenotypic females found to have histologically normal inguinal or abdominal testes. A survey in the Netherlands over a ten-year period based on reported of AIS concluded that the minimal incidence was 1:99,000 [Boehmer et al 2001].
Partial AIS (PAIS) is at least as common as complete AIS.
The prevalence of mild AIS (MAIS) has not been determined.
For current information on availability of genetic testing for disorders included in this section, see GeneTests Laboratory Directory. —ED.
Hypospadias that results from an AR gene mutation (and thus is part of the spectrum of PAIS) cannot be distinguished from hypospadias resulting from other (largely undefined) causes by the examination of the genitalia alone.
Several investigators have determined how often hypospadias of varying severity can be attributed to AR gene mutations. In studies using mutation scanning, Sutherland et al (1996) found one AR gene mutation among 40 males with penile hypospadias, while Hiort et al (1994) found no AR mutations in 12 males with coronal (glandular) or penile hypospadias but did find one person with an AR mutation among nine males with severe (penoscrotal, perineal) hypospadias. Batch et al (1992 and Batch et al (1993) found an AR mutation in two brothers with isolated severe (perineal) hypospadias. Allera et al (1995) sequenced the AR gene in nine males with isolated severe hypospadias and found an AR mutation in one. McPhaul et al (1997) found that two of the remaining eight individuals originally studied by Allera et al (1995) had androgen receptors that were transactivation deficient when their genital skin fibroblasts were infected with a recombinant adenovirus carrying an androgen-responsive reporter gene.
MAIS caused by point mutations of the AR gene [Wang et al 1998] may be clinically indistinguishable from MAIS caused by expansion of the polymorphic CAG repeat in the AR gene [Tut et al 1997]. Pathological expansion of this triplet repeat is the cause of spinobulbar muscular atrophy (SBMA), also known as Kennedy disease.
Undermasculinization of the external genitalia and pubertal undervirilization are components of many different syndromes that have no etiologic relation to the AR gene. They may or may not have a pathogenic relation to the androgen receptor protein. The one exception is a contiguous gene deletion syndrome that includes the AR gene locus and results in mental retardation and genital abnormalities [Davies et al 1997].
Findings that suggest the presence of other identifiable diagnoses in 46,XY individuals with predominantly female, ambiguous, or predominantly male genitalia include the following:
Elevated levels of testosterone precursors caused by a partial testosterone biosynthetic defect in which compensatory supranormal LH levels stimulate a normal plasma testosterone concentration
The presence of müllerian duct derivatives as a result of a testicular organogenesis defect with impaired Sertoli cell production of anti-müllerian hormone
The presence of wolffian duct-derived
internal male reproductive structures that differentiate in response to
testosterone, which suggests 5 ![]() -reductase
deficiency (However, such structures are also found in individuals with a
partial testosterone biosynthetic defect or PAIS.)
-reductase
deficiency (However, such structures are also found in individuals with a
partial testosterone biosynthetic defect or PAIS.)
Issues to consider in individuals with some, but not all, of the clinical features of AIS:
Normal plasma concentrations of T, DHT, and LH after birth do not prove that the concentration was normal during the critical period of fetal genital masculinization.
Normal responsiveness to androgen after birth does not prove that it was normal before birth. That is, early developmental delay in the acquisition of normal androgen biosynthesis or normal androgen sensitivity may simulate androgen insensitivity after birth.
Subnormal sensitivity to androgen after birth may involve components of the overall androgen response system (AR-interacting proteins) beyond the androgen receptor itself.
Evaluation of androgen receptor binding properties (i.e., binding and dissociation of specific ligands) of genital skin can help predict the likely outcome of hormone treatments (see ambiguous genitalia below).
A recent review seeks to establish a consensus statement on management of intersex disorders including AIS [Hughes et al 2006].
CAIS. A common practice is to remove the testes after puberty when feminization of the affected individual is complete, since feminization occurs partly by testicular estrogen and partly by peripheral conversion of androgen to estrogen.
The rationale for postpubertal gonadectomy is that testicular malignancy, which develops at the usual rate for cryptorchid testes, seldom occurs before puberty. Prepubertal gonadectomy is indicated if inguinal testes are physically or esthetically uncomfortable, and if inguinal herniorrhaphy is necessary. In this event, estrogen replacement therapy is necessary to initiate puberty, maintain feminization, and avoid osteoporosis.
Vaginal length may be sufficiently short to require dilatation in an effort to avoid dyspareunia.
The questions of how much to tell individuals with CAIS, and when to tell them, have not been resolved uniformly. It has become obvious, however, that systematic disclosure in an empathic setting is much preferable to systematic concealment or self-discovery of the diagnosis in an environment devoid of support from family, professionals, and other affected individuals.
PAIS with predominantly female genitalia (incomplete AIS). The issues are similar to those discussed under CAIS, except prepubertal gonadectomy helps avoid the emotional discomfort of increasing clitoromegaly at the time of puberty.
In instances in which the diagnosis of PAIS is difficult to establish because of the presence of somatic mosaicism, a change of sex assignment can result in concomitant problems [Kohler et al 2005].
PAIS with ambiguous genitalia or predominantly male genitalia. The assignment of sex in an infant with ambiguous genitalia is a complex process that requires timely assessment by a multidisciplinary team in consultation with the family and should be resolved as early as is feasible. Aside from purely anatomical and surgical considerations, the choice of a male sex-of-rearing demands a therapeutic trial with pharmacologic doses of androgen in an effort to predict potential androgen responsiveness at puberty. Furthermore, appreciable phallic growth in response to administered androgen facilitates reconstructive surgery.
In instances in which maximum information is being gathered on an infant with no family history of AIS before sex is assigned, sequence analysis of the AR gene may be considered; however, the low probability of detecting an AR mutation in individuals with the PAIS phenotype and the poor positive predictive value of any given mutation regarding AIS phenotype need to be considered when making decisions about sex assignment. The status of the androgen receptor is useful in deciding if hormone treatment could be effective and has sometimes helped in decisions about sex assignment.
Based on a small number of individuals, the role of long-term androgen pharmacotherapy in individuals with PAIS who are raised as males remains unclear. There is reason to believe that response to androgen treatment may be substantial in individuals with certain missense mutations in the DNA-binding domain of the androgen receptor [Weidemann et al 1998].
Gynecomastia that develops in puberty eventually requires reduction mammoplasty.
Those individuals with PAIS who are raised as females and who have gonadectomy after puberty may need combined estrogen and androgen replacement therapy, the latter to maintain libido.
MAIS. Men with MAIS often require reduction mammoplasty for treatment of gynecomastia.
A trial of androgen pharmacotherapy is recommended to attempt to improve virilization [Loy & Yong 2001].
The efficacy of androgen therapy in preventing such primary manifestations such as gynecomastia is not clear.
Monitoring of postnatal development of genitalia that were ambiguous at birth for changes that might lead to reconsideration of the assigned sex
For individuals assigned a male sex, evaluation during puberty for signs of gynecomastia
Search ClinicalTrials.gov for access to information on clinical studies for a wide range of diseases and conditions.
Genetic counseling is the process of providing individuals and families with information on the nature, inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members. This section is not meant to address all personal, cultural, or ethical issues that individuals may face or to substitute for consultation with a genetics professional. —ED.
AIS is inherited in an X-linked recessive manner.
Parents of a 46,XY proband
The father of a proband is not affected and is not a carrier.
Women who have an affected child and one other affected relative are obligate heterozygotes.
If a woman has more than one affected child and the disease-causing mutation cannot be detected in DNA extracted from her leukocytes, she has germline mosaicism.
If pedigree analysis reveals that the proband is the only affected family member, several possibilities exist regarding the carrier status of the proband's mother and other 46,XX females in her family.
The affected individual has a de novo AR gene mutation. There are two mechanisms by which a de novo AR gene mutation could occur:
Germline mutation. A de novo mutation was present in the egg at the time of that person's conception and is therefore present in every cell of the affected individual's body. In this instance, the individual's mother does not have an AR mutation and no other family member is at risk.
Somatic mosaicism. The mutation occurred after conception and therefore is present in some, but not all, cells of the affected individual's body [Holterhus et al 1997 , Kohler et al 2005]. In this instance the likelihood that the mother is a heterozygote is low but greater than that found in the general population.
The affected individual's mother has a de novo AR gene mutation. There are two mechanisms by which a de novo AR gene mutation could have occurred in the mother:
Germline mutation. The mutation was present in the egg or sperm at the time of her conception, is present in every cell of her body, and is detectable DNA extracted from leukocytes.
Germline mosaicism. The mutation is present only in her ovaries [Boehmer et al 1997] and is not detectable in DNA extracted from leukocytes.
Somatic mosaicism. The mutation is present in her ovaries and in some of her somatic cells and may or may not be detectable in DNA extracted from leukocytes [Kohler et al 2005].
In these instances, each of her offspring is at risk of inheriting the AR mutation; none of her sisters, however, is at risk of inheriting the AR mutation.
In 22 of 30 simplex families with CAIS or PAIS, the mother was proven to be heterozygous for an AR mutation. Of the eight individuals with de novo mutations, three appeared to have somatic mosaicism [Hiort et al 1998].
Sibs of a proband
The risk to the sibs depends upon the carrier status of the mother.
If the mother is a carrier, there is a 50% chance of transmitting the mutation to each sib.
If the proband's disease-causing mutation cannot be detected in the DNA of the mother of the only affected individual in the family, the risk to sibs is low but greater than that of the general population because the possibility of germline mosaicism exists.
Offspring of a proband. Individuals who have a 46,XY karyotype and have any of the subtypes of AIS (i.e., CAIS, PAIS, MAIS) are almost always infertile.
Offspring of a carrier female
Each offspring of a female known to be a AR mutation carrier (heterozygote) is at a 25% risk of each of the following:
The phenotype of offspring with a 46,XY karyotype and CAIS or MAIS is predictable. Although the genital phenotype of individuals with PAIS tends to vary less within a family, the wide range of phenotypes observed for the same mutation in different families makes genetic counseling difficult.
Female carriers may be identified through a combination of the following:
Clinical findings. Ten percent of carriers are manifesting carriers with asymmetric distribution and sparse or delayed growth of pubic or axillary hair, a finding that results from random X-chromosome inactivation. The presence of normal pubic and axillary hair does not rule out the possibility that an individual with a 46,XX karyotype is a carrier.
Molecular genetic testing. Carrier testing is available on a clinical basis once the AR mutation has been identified in the proband.
Androgen-binding studies on genital skin fibroblasts. In exceptional cases (i.e., no AR mutation can be identified), if an affected family member with a 46,XY karyotype is known to have deficient or defective androgen-binding activity in a genital skin fibroblast cell line, heterozygotes may be identified by the assay for deficient or defective androgen-building activity of single-cell clones from a genital skin fibroblast line.
Gender assignment. The issue of sex assignment in infancy when the child is being evaluated for ambiguous genitalia is paramount. It requires delicate decision making by parents and healthcare personnel and should be resolved as early as is feasible.
Disclosure of diagnosis. The questions of how much to tell individuals with AIS, and when to tell them, have not been resolved uniformly. It has become obvious, however, that systematic disclosure in an empathic setting is much preferable to systematic concealment or self-discovery of the diagnosis in an environment devoid of support from family, professionals, and other affected individuals [Conn et al 2005].
Family planning. The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal testing is before pregnancy.
DNA banking. DNA banking is the storage of DNA (typically extracted from white blood cells) for possible future use. Because it is likely that testing methodology and our understanding of genes, mutations, and diseases will improve in the future, consideration should be given to banking DNA of affected individuals. DNA banking is particularly relevant in situations in which the sensitivity of currently available testing is less than 100%. See DNA Banking for a list of laboratories offering this service.
Prenatal testing is possible for pregnancies of women who are carriers if an AR mutation has been identified in a family member. The usual procedure is to perform chromosome analysis on fetal cells obtained by chorionic villus sampling (CVS) at about ten to 12 weeks' gestation or by amniocentesis usually performed at about 15-18 weeks' gestation. If the karyotype is 46,XY, DNA from fetal cells can be analyzed for the known AIS-causing mutation.
Note: Gestational age is expressed as menstrual weeks calculated either from the first day of the last normal menstrual period or by ultrasound measurements.
Requests for prenatal testing for conditions such as androgen insensitivity syndrome that do not affect intellect and have some treatment available are not common. Differences in perspective may exist among medical professionals and in families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. Although most centers would consider decisions about prenatal testing to be the choice of the parents, careful discussion of these issues is appropriate.
Preimplantation genetic diagnosis (PGD)
may be available for families in which the
disease-causing mutation has been identified in an
affected family member in a research or clinical laboratory. For
laboratories offering PGD, see
![]() .
.
Information in the Molecular Genetics tables may differ from that in the text; tables may contain more recent information. —ED.
|
Gene Symbol
|
Chromosomal Locus
|
Protein Name
|
|
AR
|
Xq11-q12
|
Androgen receptor
|
|
Data are compiled from the following standard references: Gene
symbol from
HUGO;
chromosomal locus, locus name, critical region, complementation group from
OMIM; protein name from
Swiss-Prot.
|
|
|
Gene Symbol
|
Locus Specific
|
Entrez Gene
|
HGMD
|
GeneCards
|
GDB
|
GenAtlas
|
|
AR
|
|
For a description of the genomic databases listed, click
here.
|
Normal allelic variants: A polymorphic polyglutamine (CAG)n CAA tract with n=10-33 CAA repeats starts at nt 172. A polymorphic polyglycine (GGT)3 GGG(GGT)2-4 GGC(n) tract with n=4-25 starts at ~nt 1350 [Lumbroso et al 1997]. There is a silent G>A transition at the third position of codon 211 in exon 1 (according to the numerology of Lubahn et al 1988 and Lubahn et al 1989); the same codon is numbered 207 in Chang et al (1988); its nt number is 709 in Hiort et al (1994), 995 in Batch et al (1992), and 1152 in Chang et al (1988). A HindIII RFLP is detectable by a 0.7-kb fragment of the AR cDNA that extends from near the 5' border of exon 2 to about the middle of exon 7. Two polyA-addition signals occur about 220 nt apart. Coincidentally, two major species of AR mRNA (10-11 kb; ~7 kb) result from alternative splicing of a very long 3'-UTR. Two forms of the androgen receptor protein (A,B) exist. Their size difference suggests that the short form (B) represents translation initiation at the internal Met-188 residue. The issue of different forms of androgen receptor is somewhat confusing as a number of mutations that delete either whole exons or a substantial part of an exon, or affect splice sites, have been identified. In almost all cases when an attempt has been made to test these variant forms of androgen receptor protein, they have been found to be nonfunctional.
Pathologic allelic variants: Over 300 point mutations in the AR gene have been found to cause AIS [Gottlieb, Beitel, Wu, Trifiro et al 2004 ; see Androgen Receptor Gene Mutations Database]. The great majority are missense mutations that impair DNA or androgen binding and that cause CAIS or PAIS; a small number have been proven to cause MAIS. Point mutations in exon 1 are relatively infrequent. The great majority of them are either nonsense mutations or small deletions or insertions that frameshift to nonsense; hence, they almost always cause CAIS. Thus, PAIS is seldom the result of exon 1 mutations [Choong et al 1996]. A small number of major AR deletions and intronic alterations have been described.
Expansion of the polyglutamine tract to more than 40 CAG repeats causes Kennedy disease (spinobulbar muscular atrophy) with some symptoms of MAIS. (The foregoing information on disease-causing variants is found in the Androgen Receptor Gene Mutations Database [Gottlieb, Beitel, Wu, Trifiro 2004].)
Note: These conditions are not related to SBMA, as in SBMA the condition is caused by having more than 35 CAG repeats. These data involve small increases and decreases in CAG repeat length as a possible risk factor.
Normal gene product: Androgen receptor. The entire N-terminal portion of the androgen receptor (~537 aa) is encoded by exon 1, the DNA-binding domain (aa 557-616) by exons 2 and 3, the bipartite nuclear localization signal (aa 617-636) by exons 3 and 4, and the androgen-binding domain (aa 645-919) by exons 4-8. The androgen receptor is a well-defined transcriptional regulatory factor. Once activated by binding to androgen, it collaborates with other coregulatory proteins (some involve DNA binding, others do not) to achieve vectorial control over the rate of transcription of an androgen target gene that is under the influence of a nearby promoter. A large number of AR-associated proteins have now been identified [Heinlein & Chang 2002 ; Gottlieb, Beitel, Wu, Trifiro 2004]; for latest listing see the Androgen Receptor Gene Mutations Database.
Abnormal
gene product:
Nearly all
point mutations in the androgen-binding
domain impair androgen binding and, therefore, transactivation by the AR.
Some decrease only the apparent equilibrium affinity constant; some increase
only the non-equilibrium dissociation rate; others do both, either with all
androgens or selectively with certain androgens. Still others are thermolabile
or degrade excessively in the presence of androgen.
Point mutations in the zinc fingers or
![]() -helical portions of the DNA-binding
domain impair binding to a sequence of regulatory
nucleotides known as an androgen response element. Such binding is essential
for the androgen receptor to exert transcriptional regulatory control over most
of its target
genes. The polyglutamine-expanded androgen receptor causes the spinobulbar
muscular atrophy component of Kennedy disease by a gain of function that is
selectively motor neuronotoxic. The precise mechanism of its neuronotoxicity has
not been determined [Abdullah
et al 1998 ,
Beitel et al 2005].
Further, the MAIS component of Kennedy disease is caused by decreased
transcriptional regulatory activity of the polyglutamine-expanded androgen
receptor or its decreased synthesis [Beitel
et al 2005].
-helical portions of the DNA-binding
domain impair binding to a sequence of regulatory
nucleotides known as an androgen response element. Such binding is essential
for the androgen receptor to exert transcriptional regulatory control over most
of its target
genes. The polyglutamine-expanded androgen receptor causes the spinobulbar
muscular atrophy component of Kennedy disease by a gain of function that is
selectively motor neuronotoxic. The precise mechanism of its neuronotoxicity has
not been determined [Abdullah
et al 1998 ,
Beitel et al 2005].
Further, the MAIS component of Kennedy disease is caused by decreased
transcriptional regulatory activity of the polyglutamine-expanded androgen
receptor or its decreased synthesis [Beitel
et al 2005].
Recently, new insight into the relationship between mutations and their possible effects on the functionality of the actual androgen receptor protein have been obtained by creating molecular models of the receptor, using molecular dynamic modeling based on the X-ray crystal structure [Matias et al 2000]. This technique has produced dynamic models that have successfully simulated the effects of particular mutations on the ligand-binding properties of mutant androgen receptors [Elhaji et al 2004 , Wu et al 2004 , Elhaji et al 2006]. It is hoped that this technique may eventually lead to treatments that return to normal the ligand-binding capacity of mutant androgen receptors.
GeneReviews provides information about selected national organizations and resources for the benefit of the reader. GeneReviews is not responsible for information provided by other organizations. -ED.
|
![]()
No specific guidelines regarding genetic testing for this disorder have been developed.
|
|
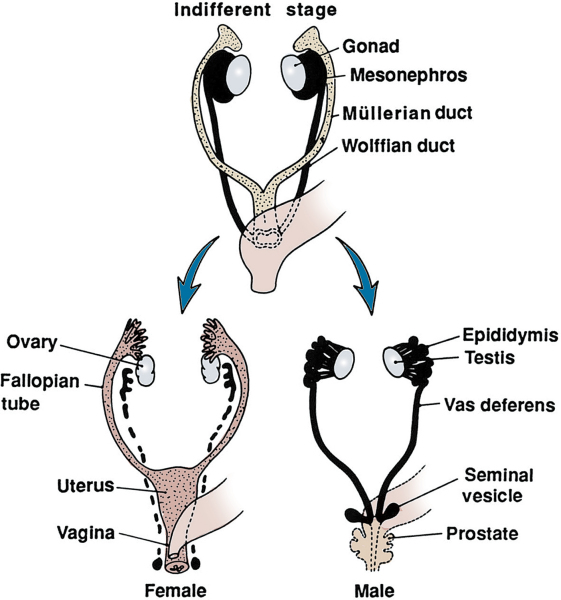
Identical structures at 8th week: differentiation apparent by10th week.
|
|
|
|
Lenore K Beitel, PhD (2004-present)
Bruce Gottlieb, PhD (2004-present)
Leonard Pinsky, MD, FACMG; McGill University (1999-2004)
Mark A Trifiro, MD (1999-present)
|
Contact GeneTests |