|
|
|
|
|
|
|
|
CHROMOSOMAL
ABNORMALITIES
|
Chromosomal anomalies occur in 0.4% of live births. They are an important cause of mental retardation and congenital anomalies. Chromosomal anomalies are present in much higher frequencies among spontaneous abortions and stillbirths. The phenotypic anomalies that result from chromosomal aberrations are mainly due to imbalance of genetic information. Chromosomal anomalies include abnormalities of chromosome number and structure.
|
ABNORMALITIES
OF CHROMOSOME
NUMBER |
When a human cell has 23 chromosomes, it is referred to as a haploid cell (the number of chromosomes in an ova or sperm). Any number of chromosomes that is an exact multiple of the haploid number (e.g., 46, 69, 92 in humans) is referred to as euploid. Euploid cells with more than the normal diploid number of 46 chromosomes are called polyploid cells. Polyploid conceptions are usually not viable. However, they may be present in mosaic (more than one cell line) forms, which allow survival. Cells with three sets of chromosomes are called triploid and are frequently seen in abortus material and occasionally in viable humans, usually in mosaic form. Cells deviating from the multiples of the haploid number are called aneuploid (i.e., not euploid), indicating a missing or extra chromosome.
The most common abnormalities of chromosome number (aneuploidy) are trisomies. These occur when there are three representatives of a particular chromosome instead of the usual two. Trisomies are usually the result of meiotic nondisjunction (failure of a chromosome pair to separate). Trisomy may be present in all cells or may occur in mosaic form. Most individuals with trisomies exhibit a consistent and specific phenotype depending on the chromosome involved ( Table 70–1 ). The most frequent trisomy in humans is trisomy 21 or Down syndrome ( Fig. 70–4 ). Trisomies of chromosome 18 ( Fig. 70–5 ) and chromosome 13 ( Fig. 70–6 ) are also relatively common and are associated with a characteristic set of congenital anomalies and mental retardation.
The incidence of Down
syndrome among conceptions is more than twice as high as it is among live
births. More than half the trisomy 21 conceptions spontaneously abort early in
pregnancy. The occurrence of trisomy 21 as well as other autosomal trisomies
increases with advancing maternal age. The increased risk of trisomy 21 in women
older than 35 yr is an indication to offer these women prenatal diagnosis.
Amniocentesis, or chorionic villus sampling to examine the fetal chromosomes, is
usually offered although maternal serum alpha fetoprotein screening and
|
TABLE 70-1 -- CHROMOSOMAL TRISOMIES AND THEIR CLINICAL FINDINGS |
|
Syndrome |
Incidence |
Clinical Manifestations |
|---|---|---|
|
Trisomy 13, Patau syndrome
|
1/10,000 births |
Cleft lip often midline; flexed fingers with polydactyly; ocular hypotelorism, bulbous nose; low-set malformed ears; small abnormal skull; cerebral malformation, especially holoprosencephaly; microphthalmia; cardiac malformations; scalp defects; hypoplastic or absent ribs; visceral and genital anomalies |
|
Trisomy 18, Edwards syndrome
|
1/6,000 births
|
Low birthweight, closed fists with index finger overlapping the 3rd digit and the 5th digit overlapping the 4th, narrow hips with limited abduction, short sternum, rocker-bottom feet, microcephaly, prominent occiput, micrognathia, cardiac and renal malformations, and mental retardation; 95% of cases are lethal in the 1st yr |
|
Trisomy 21, Down syndrome |
1/600–800 births |
Hypotonia, flat face, upward and slanted palpebral fissures and epicanthic folds, speckled irises (Brushfield spots); varying degrees of mental and growth retardation; dysplasia of the pelvis, cardiac malformations, and simian crease; short, broad hands, hypoplasia of middle phalanx of 5th finger, intestinal atresia, and high arched palate; 5% of patients with Down syndrome are the result of a translocation—t(14q21q), t(15q21q), and t(13q21q)—in which the phenotype is the same as trisomy 21 Down syndrome |
|
Trisomy 8, mosaicism |
1/20,000 births |
Long face, high prominent forehead, wide upturned nose, thick everted lower lip, microretrognathia, low-set ears, high arched, sometimes cleft palate. Osteoarticular anomalies are common; moderate mental retardation. |
|
NEURAL TUBE DEFECTS |
|
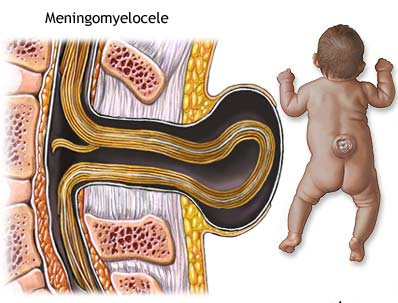
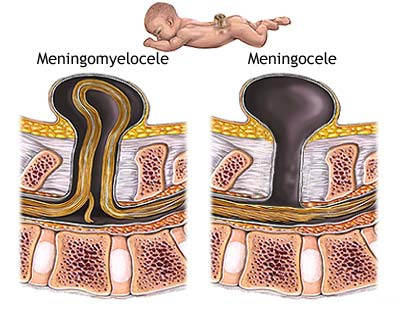
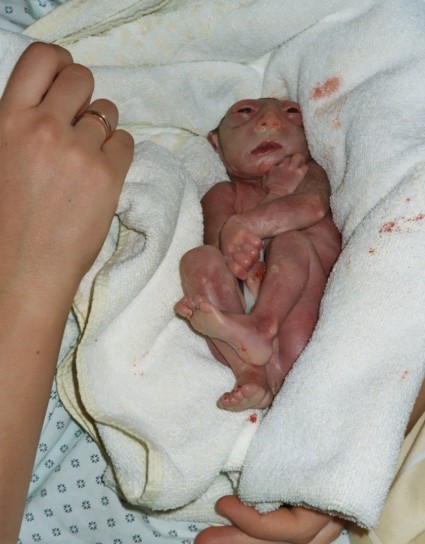
Meningomyelocele 1/800 births
|
Myelomeningocele is one of the most
common birth defects of the central nervous system.
It is a neural tube defect in which the bones of the
spine do not completely form, and the spinal canal is incomplete. This
allows the spinal cord and meninges (the membranes covering the spinal
cord) to protrude out of the child's back. Spina bifida includes any congenital defect involving insufficient closure of the spine. Myelomeningocele accounts for about 75% of all cases of spina bifida and may affect as many as 1 out of every 800 infants. The rest of the cases are most commonly spina bifida occulta (where the bones of the spine do not close, the spinal cord and meninges remain in place, and skin usually covers the defect) and meningoceles (where the meninges protrude through the vertebral defect but the spinal cord remains in place). The cause of myelomeningocele is unknown. However, folic acid deficiency is thought to play a part in neural tube defects. Also, if a child is born with myelomeningocele, subsequent children in that family have a higher risk than the general population. |
|
|
Anencephaly 4/10,000 births
|
Anencephaly is a neural tube defect that occurs early in the development of an unborn baby. Neural tube defects involve the tissue that grows into the brain and spinal cord. Anencephaly results when the upper portion of the neural tube fails to close. Why this happens is not known. Possible causes include environmental toxins and low intake of folic acid during pregnancy. Having one anencephalic infant increases the risk of having another child with neural tube defects. |
The Sunday Times - Britain
The Sunday Times May 28, 2006
Babies with club feet aborted
Lois Rogers
MORE than 20 babies have been aborted in advanced pregnancy because scans showed that they had club feet, a deformity readily corrected by surgery or physiotherapy.
According to figures from the Office for National Statistics covering the years from 1996 to 2004, a further four babies were aborted because they had webbed fingers or extra digits, which are also corrected by simple surgery. All the terminations took place late in pregnancy, after 20 weeks.
Last year, according to campaigners, a healthy baby was aborted in the sixth month at a hospital in southeast England after ultrasound images indicated part of its foot was missing.
News of the terminations has reignited the debate over how scanning and gene technology may enable the creation of “designer babies”. In 2002 it emerged that a baby had been aborted late — at 28 weeks — after scans found that it had a cleft palate, another readily corrected condition.
Some parents, doctors and charities are increasingly worried by what they see as a tendency to widen the definition of “serious handicap”. The handicap provision, which does not exist in most other countries, permits abortions to be carried out until birth. It was intended to save women from the trauma of giving birth to babies likely to die in infancy.
Club foot is one of the most common birth defects in Britain. About one in 1,000 babies is affected, meaning that 600 to 700 infants are born with the condition every year. It results in the feet pointing downwards and inwards, and in severe cases can cause foot deformity and a limp.
However, it is relatively easy to correct and in recent years techniques of splints, plaster casts and boots to set the foot into the correct position have replaced the need for surgery.Club foot is occasionally connected with serious but rare chromosomal defects, although specialists point out that these can also be screened out before birth with additional tests.
Despite the ease with which it can be treated, the perception that club foot is a serious birth defect has remained among some parents and doctors.
“It was strongly suggested that we consider abortion after they found our baby had a club foot,” said David Wildgrove, 41, a computer programmer from Sheffield, whose son Alexander was born in 1996. “I was appalled. We resisted, the problem was treated and he now runs around and plays football with everyone else.”
Pippa Spriggs from Cambridge, whose son Isaac will celebrate his second birthday in July, was also dismayed when a scan halfway through the pregnancy revealed that her baby had the defect.
“Abortion certainly was not openly advised, but it was made clear to me it was available,” she said. “In fact he has been treated and the condition has not slowed him down at all.”
Others take a different view and decide not to accept the risk of an imperfect baby. Sue Banton, who founded the group Steps for parents of children with foot disorders, was troubled that a home counties couple last year decided to terminate their baby, despite counselling to reassure them it would have a worthwhile life even with a section of foot missing.
“We gave them other families to talk to, but they just didn’t want to know. The baby was aborted just before the 25th week,” she said.
“It is terrible. I know lots of perfectly nice people with this condition, and you just can’t imagine them not being here.”
One doctor in the north of England who did not want to be named, said a recent case in his hospital had involved the discovery of a hand missing from a foetus scanned at 20 weeks. “The father did not want the pregnancy to proceed because of his perception that the child would not be able to do all the usual things like sport,” said the doctor.
Naomi Davis, a leading paediatric surgeon at Manchester children’s hospital who specialises in correcting club feet, said: “I think it’s reasonable to be totally shocked that abortion is being offered for this. It is entirely treatable. I can only think it is lack of information.”
Jane Fisher, director of the charity Antenatal Results and Choices, defended the right of parents to terminate pregnancies when defects were found.
“This is not part of a move towards designer babies,” she said. “These are difficult and painful issues.”
Club foot has been no bar to high achievement. Eminent sufferers have included:
Lord Byron, the poet, who swam the mile across the Hellespont and wrote a verse about it
Dudley Moore, the actor and musician oDavid Starkey, the television historian and political critic
Toyah Willcox, the former chart-topping singer, who was born with a twisted spine as well as a club foot
Kristi Yamaguchi, an American figure skater who won an Olympic gold medal in 1992



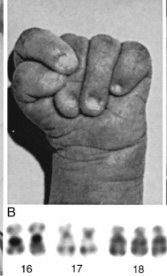
 Figure 70-4
Partial karyotypes from patients with Down syndrome. A, Patient with
trisomy 21. B, Chromosome 21 from two patients and their parents.
Left: Two of a patient's chromosomes with brightly fluorescent satellites
were transmitted by the mother. Right: Another patient's two chromosomes
with bright satellites resulted from paternal nondisjunction at second meiotic
division. C, 21q21q translocation. D, 14q21q translocation in a mother (above)
and her affected child (below).
Figure 70-4
Partial karyotypes from patients with Down syndrome. A, Patient with
trisomy 21. B, Chromosome 21 from two patients and their parents.
Left: Two of a patient's chromosomes with brightly fluorescent satellites
were transmitted by the mother. Right: Another patient's two chromosomes
with bright satellites resulted from paternal nondisjunction at second meiotic
division. C, 21q21q translocation. D, 14q21q translocation in a mother (above)
and her affected child (below).
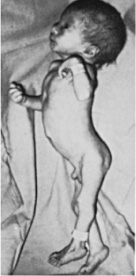
 Figure 70-5
A, Photograph of male infant with trisomy 18, age 4 days. Note prominent
occiput, micrognathia, low-set ears, short sternum, narrow pelvis, prominent
calcaneus, and flexion abnormalities of the fingers. (Courtesy of Robert E.
Carrel.) B, Several of the common anomalies in the 18 trisomy syndrome,
including the unusual position of the fingers with hypoplasia of fifth
fingernail, the simple arch pattern of the finger pads, and the dorsiflexed
hallux with hypoplasia of toenails. (From Smith DW: Autosomal abnormalities.
Am J Obstet Gynecol 1964;90:1055.) C, Partial karyotype of trisomy 18
prepared with modified Giemsa stain.
Figure 70-5
A, Photograph of male infant with trisomy 18, age 4 days. Note prominent
occiput, micrognathia, low-set ears, short sternum, narrow pelvis, prominent
calcaneus, and flexion abnormalities of the fingers. (Courtesy of Robert E.
Carrel.) B, Several of the common anomalies in the 18 trisomy syndrome,
including the unusual position of the fingers with hypoplasia of fifth
fingernail, the simple arch pattern of the finger pads, and the dorsiflexed
hallux with hypoplasia of toenails. (From Smith DW: Autosomal abnormalities.
Am J Obstet Gynecol 1964;90:1055.) C, Partial karyotype of trisomy 18
prepared with modified Giemsa stain.

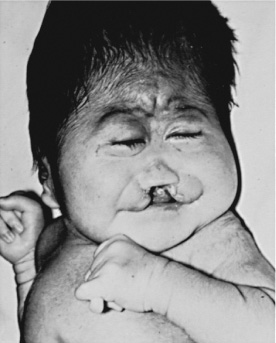
 Figure 70-6
A and B, Female infants with trisomy 13 syndrome. Note the midline
cleft of the lip and palate, microcephaly, hypotelorism, microphthalmos, bulbous
nose, polydactyly, and overlapping of fingers. Scalp defects (not shown) are
also present. (Courtesy of Miriam G. Wilson.) C, Partial karyotype
showing chromosomes 13, 14, and 15 stained with the trypsin-Giemsa method.
Figure 70-6
A and B, Female infants with trisomy 13 syndrome. Note the midline
cleft of the lip and palate, microcephaly, hypotelorism, microphthalmos, bulbous
nose, polydactyly, and overlapping of fingers. Scalp defects (not shown) are
also present. (Courtesy of Miriam G. Wilson.) C, Partial karyotype
showing chromosomes 13, 14, and 15 stained with the trypsin-Giemsa method.
recovering fetal DNA or cells from maternal blood are increasingly being used. In women younger than 35 yr of age, maternal serum testing (triple screen) can be efficacious in prenatal screening for Down syndrome. Low maternal serum α-fetoprotein concentration, low unconjugated estriol, and elevated human chorionic gonadotropin are indicators of Down syndrome. Prenatal ultrasonography may detect a thickened nuchal fold, absent nasal bone, a shortened femur, and cardiac or gastrointestinal anomalies associated with trisomy 21 ( Chapter 85 ).
All individuals with Down syndrome have three copies of chromosome 21. About 95% have three freestanding copies of chromosome 21. Approximately 1% of individuals are mosaic with some normal cells. Approximately 4% of Down syndrome individuals have a translocation involving chromosome 21. Translocations account for 9% of the children with Down syndrome born to mothers younger than 30 yr of age. Half the translocations arise de novo in the affected individual, whereas half are inherited from a translocation carrier parent. Parents who are carriers of a translocation involving chromosome 21 produce three types of viable offspring: normal phenotype and karyotype, a phenotypically normal translocation carrier, and the translocation trisomy 21. The majority of translocations that give rise to Down syndrome are fusions at the centromere between chromosomes 13, 14, 15, or 21, for example, t(13q,15q) or t(21q,21q). The phenotype in translocation Down syndrome is not distinguishable from regular trisomy 21 Down syndrome (see Table 70–1 ). Chromosome studies must be performed on every Down syndrome individual. If a translocation is identified, parental studies must be performed to identify normal individuals who are translocation carriers with a high recurrence risk for a chromosomally abnormal child and who may also have other family members at risk.
Monosomies occur when only one representative of a chromosome is present. They may be complete or partial. Complete monosomies may be the result of nondisjunction or anaphase lag. In nondisjunction during cell division, the two chromosomes in a replicating pair fail to separate; one cell ends up with only one copy (monosomic) and the other with three copies (trisomic) of the specific chromosome. In anaphase lag, a chromosome fails to move into the new daughter cell and is lost. In humans, all complete autosomal monosomies appear to be lethal early in development and only survive in mosaic forms. Partial monosomies are usually the offspring of a translocation carrier.
Deletions occur when a piece of a chromosome is missing. They may occur as a simple deletion or as a deletion with duplication of another chromosome segment. The latter is usually caused by a crossover in meiosis in a translocation carrier, resulting in an unbalanced reciprocal chromosomal translocation. Deletions may be located at the chromosome ends or in interstitial segments of the chromosome and are usually associated with mental retardation and malformations. Small telomeric deletions may be relatively common in nonspecific mental retardation with minor anomalies. The most commonly observed deletions in humans are 4p-, 5p-, 9p-, 11p-, 13q-, 18p-, and 18q-, which are associated with well-described phenotypes ( Table 70–2 ). Deletions may be observed in routine chromosome preparations, but microdeletions are detectable only under the microscope with prophase chromosome studies. In submicroscopic deletions, the missing piece can be detected only by using molecular probes or DNA studies.
Microdeletions are defined as small chromosome deletions that are detectable only in high-quality (pro)metaphase preparations. These deletions often involve several genes so that the affected individual may be identified by an unusual phenotype associated with an apparent single gene mutation. Williams, Langer-Giedion,
|
Deletion |
Clinical Abnormalities |
|---|---|
|
4p- |
Wolf-Hirschhorn syndrome. The main features are a typical “Greek helmet” facies with ocular hypertelorism, prominent glabella, and frontal bossing; microcephaly, dolichocephaly, hypoplasia of the eye socket, ptosis, strabismus, nystagmus, bilateral epicanthic folds, cleft lip and palate, beaked nose with prominent bridge, hypospadias, cardiac malformations, and mental retardation. |
|
5p- |
Cri-du-chat syndrome. The main features are hypotonia, short stature, characteristic cry, microcephaly with protruding metopic suture, moonlike face, hypertelorism, bilateral epicanthic folds, high arched palate, wide and flat nasal bridge, and mental retardation. |
|
9p- |
The main features are craniofacial dysmorphology with trigonocephaly, slanted palpebral fissures, discrete exophthalmos, arched eyebrows, flat and wide nasal bridge, short neck with pterygium colli, genital anomalies, long fingers and toes, cardiac malformations, and mental retardation. |
|
13q- |
The main features are low birthweight, failure to thrive, and severe mental retardation. Facial features include microcephaly, flat wide nasal bridge, hypertelorism, ptosis, micrognathia. Ocular malformations are common. The hands have hypoplastic or absent thumbs and syndactyly. |
|
18p- |
A few patients (15%) are severely affected and have cephalic and ocular malformations, cleft lip and palate, and varying degrees of mental retardation. Most (80%) have only minor malformations and mild mental retardation. |
|
18q- |
The main features are hypotonia with “froglike” position with the legs flexed, externally rotated, and in hyperabduction. The face is characteristic with depressed midface and apparent protrusion of the mandible, deep-set eyes, short upper lip, everted lower lip (“carplike” mouth); antihelix of the ears is very prominent; varying degrees of mental retardation and belligerent personality. |
|
21q- |
The main features are hypertonia, microcephaly, downward-slanting palpebral fissures, high palate, prominent nasal bridge, large low-set ears, micrognathia, and varying degrees of mental retardation. They may have skeletal malformations. |
Prader-Willi, Angelman, Rubinstein-Taybi, Smith-Magenis, Miller- Dieker,
Alagille, and velocardiofacial/DiGeorge syndromes have all been found to be
associated with microdeletions ( Table 70–3 ).
Submicroscopic deletions are not visible by microscopic examination and are
detected only with specific probes for a DNA sequence or DNA studies. The
deletion is recognized because of the absence of staining or fluorescence.
Translocations involve the transfer of chromosomal material from one chromosome to another. Translocations may be Robertsonian or reciprocal. They occur with a frequency of 1/500 liveborn human infants. They may be inherited from a parent or appear de novo, with no other affected family members.
Robertsonian translocations involve two acrocentric (centromere located at the end) chromosomes that fuse near the centromeric region with subsequent loss of the nonfunctional, very truncated short arms. The translocation chromosome is made up of the long arms of two fused chromosomes, hence the resulting count is only 45 chromosomes. The loss of the short arms of acrocentric chromosomes has no known deleterious effect. Although carriers of a Robertsonian translocation are usually phenotypically normal, they are at increased risk for miscarriages and abnormal offspring. Reciprocal translocations are the result of breaks in nonhomologous chromosomes with reciprocal exchange of the broken segments. Carriers of a reciprocal translocation are usually phenotypically normal but also have an increased risk of having chromosomally abnormal offspring and miscarriages because of abnormalities in the segregation of the chromosomes in the germ cells.