|
Hwang Woo-Suk |
|
|
|
Hwang Woo-Suk |
|
|
Science, Vol 303, Issue 5664, 1669-1674, 12 March 2004
EVIDENCE of a PLURIPOTENT HUMAN EMBRYONIC STEM CELL LINE DERIVED from a CLONED BLASTOCYST
Woo Suk Hwang,1,2* Young June Ryu,1 Jong Hyuk Park,3 Eul Soon Park,1 Eu Gene Lee,1 Ja Min Koo,4 Hyun Yong Chun,1 Byeong Chun Lee,1 Sung Keun Kang,1 Sun Jong Kim,3 Curie Ahn,5 Jung Hye Hwang,6 Ky Young Park,7 Jose B. Cibelli,8 Shin Yong Moon5*
1College of Veterinary Medicine, Seoul National University, Seoul 151-742, Korea. 2School of Agricultural Biotechnology, Seoul National University, Seoul 151-742, Korea. 3Medical Research Center, Mizmedi Hospital, Seoul, 135-280, Korea. 4Gachon Medical School, Incheon, 417-840, Korea. 5College of Medicine, Seoul National University, Seoul, 110-744, Korea. 6School of Medicine, Hanyang University, Seoul, 471-701, Korea. 7College of Natural Science, Sunchon National University, Sunchon, 540-742, Korea. 8Department of Animal Science-Physiology, Michigan State University, East Lansing, MI 48824, USA.
Somatic cell nuclear transfer (SCNT) technology has recently been used to generate animals with a common genetic composition. In this study, we report the derivation of a pluripotent embryonic stem cell line (SCNT-hES-1) from a cloned human blastocyst. SCNT-hES-1 cells display typical ES cell morphology and cell surface markers and are capable of differentiating into embryoid bodies in vitro and of forming teratomas in vivo containing cell derivatives from all three embryonic germ layers in SCID mice. After continuous proliferation for >70 passages, SCNT-hES-1 cells maintain normal karyotypes and are genetically identical to the somatic nuclear donor cells. Although we cannot completely exclude the possibility of a parthenogenetic origin of the cells, imprinting analyses provide support that the derived human ES cells have a somatic cell nuclear transfer origin.
The isolation of pluripotent human embryonic stem (ES) cells (1) and breakthroughs in somatic cell nuclear transfer (SCNT) in mammals (2) have raised the possibility of performing human SCNT to generate potentially unlimited sources of undifferentiated cells for research, with potential applications in tissue repair and transplantation medicine. This concept, known as “therapeutic cloning,” refers to the transfer of the nucleus of a somatic cell into an enucleated donor oocyte (3). In theory, the oocyte’s cytoplasm would reprogram the transferred nucleus by silencing all the somatic cell genes and activating the embryonic ones. ES cells would be isolated from the inner cell mass (ICM) of the cloned preimplantation embryo. When applied in a therapeutic setting, these cells would carry the nuclear genome of the patient; therefore, it is proposed that following directed cell differentiation, the cells could be transplanted without immune rejection for treatment of degenerative disorders such as diabetes, osteoarthritis, and Parkinson’s disease, among others. Previous reports have described the generation of bovine ES-like cells (4) and mouse ES cells from ICMs of cloned blastocysts (5–7), and the development of cloned human embryos to 8 to 10 cell stage (8, 9). Here we describe evidence of the derivation of human ES cells after SCNT (10).
Fresh oocytes and cumulus cells were donated by healthy women for the express purpose of SCNT stem cell derivation for therapeutic cloning research and its applications. Prior to beginning any experiments, we obtained approval for this study from the Institutional Review Board on Human Subjects Research and Ethics Committees (Hanyang University Hospital, Seoul, Korea). Donors were fully aware of the scope of our study and signed an informed consent form (supporting online text); donors voluntarily donated oocytes and cumulus cells (including DNA) for therapeutic cloning research and its applications only, not for reproductive cloning; there was no financial payment. A total of 242 oocytes were obtained from 16 volunteers (one or two donors for each trial) after ovarian stimulation and 176 metaphase II (MII) oocytes were used for SCNT. Autologous SCNT was performed, i.e. the donor's own cumulus cell, isolated from the cumulus-oocyte-complex (COC), was transferred back into the donor's own enucleated oocyte. Prior to enucleation, the oocytes were matured in vitro in G1.2 medium (Vitro Life, Goteborg, Sweden) for 1 to 2 hrs. Enucleation, SCNT, and electrical fusion were performed as described (11). To confirm directly that the oocyte’s DNA was removed during enucleation, we imaged the extruded DNA-MII spindle complex from every oocyte with Hoechst 33342 fluorescent DNA dye (Fig. 1, A and B: arrows).
Absent any report of an efficient protocol for human SCNT, several critical steps had to be optimized (2), including reprogramming time, activation method and in vitro culture conditions. Reprogramming time, or the lapse of time between cell fusion and egg activation, returns the gene expression of the somatic cell to that needed for appropriate embryonic development. Initially, we investigated the effect of simultaneous fusion and activation, as used for porcine SCNT (12, 13), but observed low fusion and cleavage rates, with no blastocyst development. Instead, we adapted the bovine SCNT procedure of waiting a few hours between fusion and activation. By allowing two hours for reprogramming, we were able to obtain ~25% of the embryos to the blastocyst stage. Since sperm-mediated activation is absent in SCNT, an artificial stimulus is needed to initiate development. Various chemical, physical, and mechanical agents induce parthenogenetic development in mice (14), but human data are limited.
Oocyte activation using the calcium ionophore A23187 (calcimycin) or ionomycin and the protein synthesis inhibitor puromycin induces parthenogenetic development of human oocytes at different efficiencies (15). We found that incubation in 10 µM A23187 for 5 min followed by incubation with 2.0 mM 6-dimethylaminopurine (DMAP) for 4 hrs gave efficient chemical activation for human SCNT eggs. Other investigators have reported encouraging results in overcoming inefficiencies in embryo culture by supplementing with different energy substrates (16). Furthermore, recent development of serum-free sequential media has led to considerable improvements in the rate of clinical pregnancies (17). In this study, the human modified synthetic oviductal fluid (SOF) with amino acids (hmSOFaa) was prepared by supplementing mSOFaa (18) with human serum albumin (10 mg/ml) and fructose (1.5 mM) instead of bovine serum albumin (8 mg/ml) and glucose (1.5 mM), respectively. The replacement of glucose with fructose improves the developmental competence of bovine SCNT embryos (11, 19). Culture of human SCNT-derived embryos in G1.2 medium for the first 48 hrs followed by hmSOFaa medium produced more blastocysts, compared to G1.2 medium for the first 48 hrs followed by culture in G1.2 medium or in continuous hmSOFaa medium (Table 1). Cibelli et al. (8) reported that the treatment of human oocytes with 5 µM calcium ionomycin followed by 2 mM DMAP in G1.2 culture medium triggered pronucleus formation, embryonic cleavage, and the formation of a blastocoelic cavity in human parthegenotes. However, they did not obtain human SCNT blastocysts when their protocol was applied to SCNT embryos. Oocyte limitations precluded full optimization of all the parameters for human SCNT; nonetheless, the protocol described here produced cloned blastocysts at rates of 19 to 29% (as a percentage of reconstructed eggs) and was comparable to those from established SCNT methods in cattle (~25%) (11) and pigs (~26%) (12, 13).
[1] A total of 30 SCNT-derived blastocysts were cultured,
[2] 20 ICMs were isolated by immunosurgical removal of the trophoblast, and
[3] one ES cell line (SCNT-hES-1) was derived.
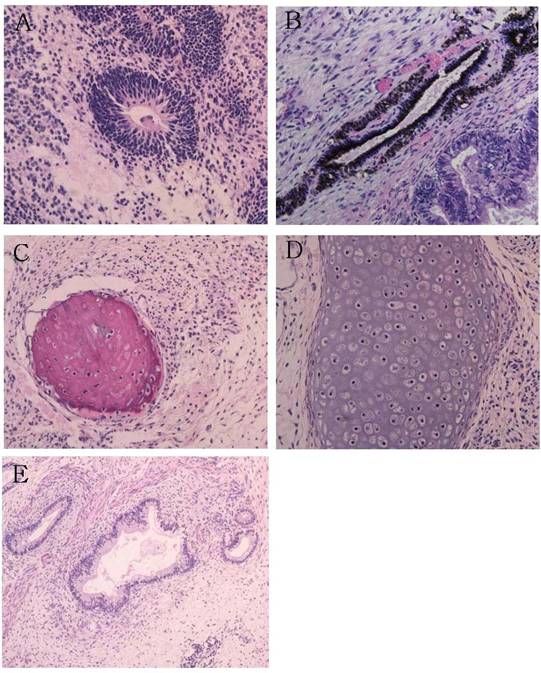
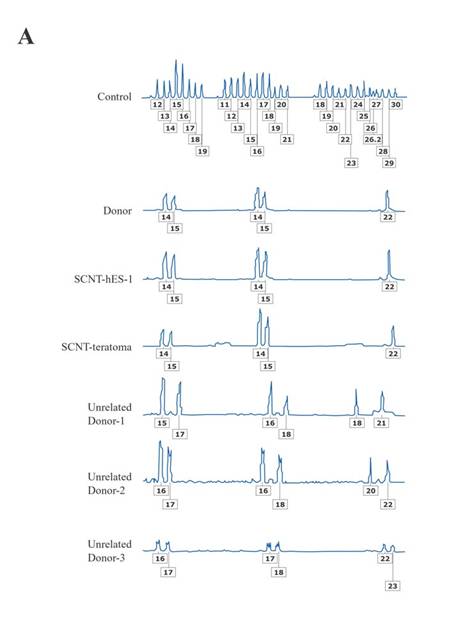
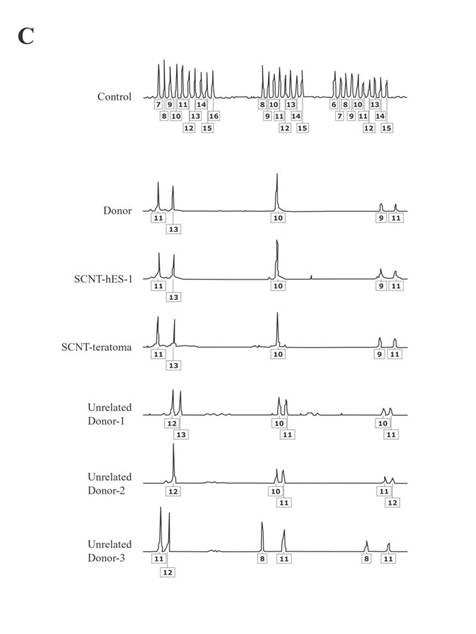
The resulting SCNT-hES-1 cells had a high nucleus to cytoplasm ratio and prominent nucleoli. The cell colonies display similar morphology to that reported previously for hES cells derived after IVF (Fig. 1, C to E). When cultured in defined medium conditioned for neural cell differentiation (20), SCNT-hES-1 cells differentiated into nestin positive cells, an indication of primitive neuroectoderm differentiation (Fig. 1F). The SCNT-hES-1 cell line was mechanically passaged by dissociation every five to seven days and successfully maintained its undifferentiated morphology after continuous proliferation for > 70 passages, while still maintaining a normal female (XX) karyotype (Fig. 1G) (21). Furthermore, SCNT–hES-1 cells express ES cell markers such as alkaline phosphatase, SSEA-3, SSEA-4, TRA-1-60, TRA-1-81, and Oct-4, but not SSEA-1 (Fig. 2). As previously described in monkey (22) and human ES cells (1, 23, 24), and mouse SCNT-ES cells (6), SCNT-hES-1 cells do not respond to exogenous leukaemia inhibitory factor (LIF), suggesting that a pluripotent state is maintained by a gp130 independent pathway. Pluripotency of SCNT-hES-1 cells was investigated in vitro (fig. S1) and in vivo (Fig. 3). Clumps of the cells were cultured in vitro in suspension to form embryoid bodies. The resulting embryoid bodies were stained with three dermal markers and were found to differentiate into a variety of cell types including derivatives of endoderm, mesoderm, and ectoderm (fig. S1). When undifferentiated SCNT-hES-1 cells were injected into the testis of SCID mice, teratomas were obtained from six to seven weeks after injection. The resulting teratomas contained tissue representative of all three germ layers. Differentiated tissues seen in Fig. 3 include neuroepithelial rosset, pigmented retinal epithelium, smooth muscle, bone, cartilage, connective tissues, and glandular epithelium. The DNA fingerprinting analysis with human short tandem repeat (STR) markers indicates that the cell line originated from the cloned blastocysts reconstructed from the donor cells, not from parthenogenetic activation (Fig. 4, A to C). The statistical probability that the cells may have derived from an unrelated donor is 8.8 × 10–16. The RT-PCR amplification for paternally-expressed (hSNRPN and ARH1) and maternally-expressed (UBE3A and H19) genes further confirmed that the cell line originated from the donor cells (Fig. 4D).
Simerly et al. (26) recently reported defective mitotic spindles after SCNT in nonhuman primate embryos, perhaps resulting from the depletion of microtubule motor and centrosome proteins lost to the meiotic spindle after enucleation. In this study, Fig. 1G demonstrates that SCNT-hES-1 cells have the normal karyotype. We speculate that other SCNT-blastocysts from which ES cell lines were not derived might have been aneuploid. However, it is important to note that our investigations differ from Simerly et al. in a few ways: media and protocols for in vitro development have been optimized for human oocytes and embryos, whereas the protocols for nonhuman primate studies are extrapolated from the clinical procedures; the enucleation method here differs since we squeeze the MII oocyte so that the DNA-spindle complex is extruded through a small hole in the zona pellucida, instead of aspirating the DNA-spindle complex with a glass pipette as others have described (27); and the DNA-spindle complex is extruded shortly after the appearance of the first polar body, so that it may even be at the prometaphase II stage.
In this report, we provide three lines of evidence supporting the NT origins of SCNT-hES-1: 1) DNA extraction was verified for each of the 242 enucleated oocytes [Fig. 1, A and B (arrows)]; 2) DNA fingerprinting shows heterozygous, not homozygous, chromosomes (Fig. 4, A to C); and 3) biparental, and not unimaternal, expression of imprinted genes (Fig. 4D). Whereas the Cyno1 parthenogenetic cells retain their strictly maternal imprints, that evidence comes from a single monkey cell line. Given the aberrant expression of imprinted genes after murine SCNT (28), perhaps SCNT-hES-1 cell’s biparental expression of imprinted genes might have been influenced by SCNT or subsequent culture. Heterologous along with autologous SCNT will provide more definitive molecular evidence. Whereas overwhelming ethical constraints preclude any reproductive cloning attempts, complementary investigations in nonhuman primates might provide additional, confirmatory information. Consequently, while we cannot exclude the possibility of a parthenogenetic origin, the studies reported here support the conclusion that the SCNT-hES-1 cell line originated from the donor's diploid somatic cumulus cell after SCNT.
In order to successfully derive immuno-compatible human ES cells from a living donor, a reliable and efficient method for producing cloned embryos and ES isolation must be developed.
homson et al. (1), Reubinoff et al. (23), and Lanzendorf et al. (29) produced human E[mbryonic]S[tem] cell lines at high efficiency. [these researchers dissected normally-fertilized human embryos] Briefly, five ES cell lines were derived from a total of 14 ICMs, two ES cell lines from four ICMs, and three ES cell lines from 18 ICMS, respectively.
In our study, one S[omatic]C[cell]N[uclear]T[ransfer]-hES cell line was derived from 20 ICMs.
It remains to be determined if this low efficiency is due to faulty reprogramming of the somatic cells or subtle variations in our experimental procedures. We cannot rule out the possibility that the genetic background of the cell donor had an impact on the overall efficiency of the procedure. Further improvement in SCNT protocols and in vitro culture systems are needed before contemplating the use of this technique for cell therapy. In addition, those mechanisms governing the differentiation of human tissues must be elucidated in order to produce tissue-specific cell populations from undifferentiated ES cells. This study shows the feasibility of generating human embryonic stem cells from a somatic cell isolated from a living person.
References and Notes
1. 1. J. A. Thomson et al., Science 282, 1145 (1998).
2. 2. D. Solter, Nat. Rev. Genet. 1, 199 (2000).
3. 3. R. P. Lanza, J. B. Cibelli, M. D. West, Nat. Med. 5, 975 (1999).
4. 4. J. B. Cibelli et al. Nat. Biotechnol. 16, 642 (1998).
5. 5. M. J. Munsie et al., Curr. Biol. 10, 989 (2000).
6. 6. E. Kawase, Y. Yamazaki, T. Yagi, R. Yanagimachi, R. A. Pederson, Genesis 28, 156 (2000).
7. 7. T. Wakayama et al., Science 292, 740 (2001).
8. 8. J. B. Cibelli et al., J. Reg. Med. 26, 25 (2001).
9. 9. Y. Shu, G. Zhuang, Fertil. Steril. 78, S286 (2002).
10. 10. Materials and methods are available as supporting material on Science Online.
11. 11. J. Kwun et al., Mol. Reprod. Dev. 65, 167 (2003).
12. 12. S. H. Hyun et al., Biol. Reprod. 69, 1060 (2003).
13. 13. B. Kuhholzer, R. J Hawley, L. Lai, D. Kolber-Simonds,
R. S. Prather, Biol. Reprod. 64, 1635 (2004).
1. 14. M. H. Kaufman, Nature 242, 475 (1973).
2. 15. K. Nakagawa et al., Zygote 9, 83 (2001).
3. 16. D. K. Gardner, M. Lane, W. B. Schoolcraft, J. Reprod. Immunol. 55, 85 (2002).
4. 17. A. Langendonckt, D. Demylle, C. Wyns, M. Nisolle, J. Donnez, Fertil. Steril. 76, 1023 (2001).
5. 18. Y. H. Choi, B. C. Lee, J. M. Lim, J. M. S. K Kang, W. S. Hwang, Theriogenology 58, 1187 (2002).
6. 19. D. K. Barnett, B. D. Bavister, Mol. Reprod. Dev. 43, 105 (1996).
7. 20. S. H. Lee, N. Lumelsky, L. Studer, J. M., Auerbach, R. D. Mckay, Nat. Biotech. 18, 675 (2000).
8. 21. F. Mitelman, An International System For Human Cytogenetic Nomenclature (S. Karger Publisher, Basel, Switzerland, 1995).
22. J. A. Thomson et al., Proc. Natl. Acad. Sci.U.S.A. 92, 7844 (1995).
1. 23. B. E. Reubinoff, F. P. Pera, C.-Y. Fong, A. Trounson, A. Bongso, Nat. Biotech. 18, 399 (2000).
2. 24. S. R. John, Trends Biotech. 20, 417 (2002).
3. 25. K. E. Vrana et al., Proc. Natl. Acad. Sci. U.S.A. 100 Suppl. 1, 1911 (2003).
4. 26. C. Simerly et al., Science, 300, 297 (2003).
5. 27. I. Wilmut et al., Nature 385, 810 (1997).
28. D. Humphreys et al., Proc. Natl. Acad. Sci. U.S.A. 99, 12889 (2002).
1. 29. S. E. Lanzendorf et al., Fertil. Steril. 76, 132 (2001).
2. 30. We thank Y. Y Hwang (Hanyang University) for assistance with oocyte collections; S. I. Rho (Mizmedi Hospital), H. S. Yoon (Mizmedi Hospital) and S. K. Oh (Seoul National University) for assistance on hES cells culture; Y. K. Choi (KRIBB) for assistance on teratoma formation; Tak Ko (Michigan State University) for gene expression analysis of Cyno-1 cells; and A. Trounson (Monash University), B. D. Bavister (University of New Orleans) and D. P. Wolf (Oregon National Primate Research Center) for critical review of the manuscript. J.B. Cibelli has made intellectual contributions to the manuscript and RNA analysis of nonhuman primate cells. All human experiments were performed in Korea by Korean scientists. This study was supported by grants from the Advanced Backbone IT Technology Development (IMT2000-C1-1) to W.S.H. and the Center for Stem Cell Research (M102KL0100-02K1201-00223) to S.Y.M.. The authors are grateful for a graduate fellowship provided by the Ministry of Education, through the BK21 program.
Supporting Online Material
www.sciencemag.org/cgi/content/full/1094515/DC1 Materials and Methods SOM Text Fig. S1
9 December 2003; accepted 4 February 2004 Published online 12 February 2004; 10.1126/science.1094515 Include this information when citing this paper.
![]()
Fig. 4. The DNA fingerprinting analysis and expression of imprinted genes. (A) Isogenic analysis in loci D3S1358 (chromosome location: 3p), loci vWA (chromosome location: 12p12-pter), and loci FGA (chromosome location: 4q28). (B) Isogenic analysis in loci amelogenin (Chromosome location: X:p22.1-22.3, Y:p11.2), loci THO1 (chromosome location: 11p15.5), loci TPOX (chromosome location: 2p23-2per), and loci CSF1PO (chromosome location: 5q33.3-34). (C) Isogenic analysis in loci D5S818 (chromosome location: 5p22-31), loci D13S317 (chromosome location: 13q22-31), and loci D7S820 (chromosome location: 7q11.21-22). The boxed numbers and corresponding peaks represent locations of polymorphisms for each short tandem repeat marker. (D) The RT-PCR amplification of paternally-expressed (hSNRPN and ARH1) and maternally-expressed (UBE3A and H19) genes. Cyno-1, maternally-derived monkey parthenogenic stem cell line (25); mFBLST; monkey fibroblasts; hFBLST, human fibroblasts; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Tm(-), without template for PCR amplification.
Table 1.
Conditions for human somatic cell nuclear transfer.
|
Experiment |
Activation condition* |
Reprogramming time (hrs) |
1st step medium† |
2nd step medium |
No. of oocytes |
No. (%) of cloned embryos developed to |
|||
|
2-cell |
Compacted morula |
Blastocyst |
|||||||
|
|
10 µM ionophore |
6-DMAP |
2 |
G 1.2 |
hmSOFaa |
16 |
16 (100) |
4 (25) |
4 (25) |
|
1st set |
10 µM ionophore |
6-DMAP |
4 |
G 1.2 |
hmSOFaa |
16 |
15 (94) |
1 (6) |
0 |
|
10 µM ionophore |
6-DMAP |
6 |
G 1.2 |
hmSOFaa |
16 |
15 (94) |
1 (6) |
1 (6) |
|
|
|
10 µM ionophore |
6-DMAP |
20 |
G 1.2 |
hmSOFaa |
16 |
9 (56) |
1 (6) |
0 |
|
|
10 µM ionophore |
6-DMAP |
2 |
G 1.2 |
hmSOFaa |
16 |
16 (100) |
5 (31) |
3 (19) |
|
2nd set |
5 µM ionophore |
6-DMAP |
2 |
G 1.2 |
hmSOFaa |
16 |
11 (69) |
0 |
0 |
|
10 µM ionomycin |
6-DMAP |
2 |
G 1.2 |
hmSOFaa |
16 |
12 (75) |
0 |
0 |
|
|
|
5 µM ionomycin |
6-DMAP |
2 |
G 1.2 |
hmSOFaa |
16 |
9 (56) |
0 |
0 |
|
|
10 µM ionophore |
6-DMAP |
2 |
G 1.2 |
hmSOFaa |
16 |
16 (100) |
4 (25) |
3 (19) |
|
3rd set |
10 µM ionophore |
6-DMAP |
2 |
G 1.2 |
G 2.2 |
16 |
16 (100) |
0 |
0 |
|
|
10 µM ionophore |
6-DMAP |
2 |
Continuous hmSOFaa |
16 |
16 (100) |
0 |
0 |
|
|
4th set |
10 µM ionophore |
6-DMAP |
2 |
G 1.2 |
hmSOFaa |
66 |
62 (93) |
24 (36) |
19 (29) |
*Fused donor oocytes and somatic cells were activated in either calcium ionophore A23187 (5 or 10 µM) or ionomycin (5 or 10 µM) for 5 min, followed by 2 mM 6-dimethylaminopurine (DMAP) treatment for 4 hrs. †Oocytes were incubated in first medium for 48 hrs.
Fig. 1. Confirmation of enucleation, photographs of human SCNT ES cells and their differentiated progeny, and karyotype analysis. Images (×200) of extruded DNA-MII spindle complexes (arrows) from oocyte before (A) and after enucleation (B). The bright-field [(C), ×100] and phase contrast [(D), ×100] micrographs, and higher magnification [(E), ×200] of a colony of SCNT-hES-1 cells. Immunofluorescence staining for nestin [(F), ×200] and the G-banded kayotyping (G) in SCNT-hES-1 cells. Scale bars = 20 µm [(A) and (B)] and 100 µm [(C) to (F)].
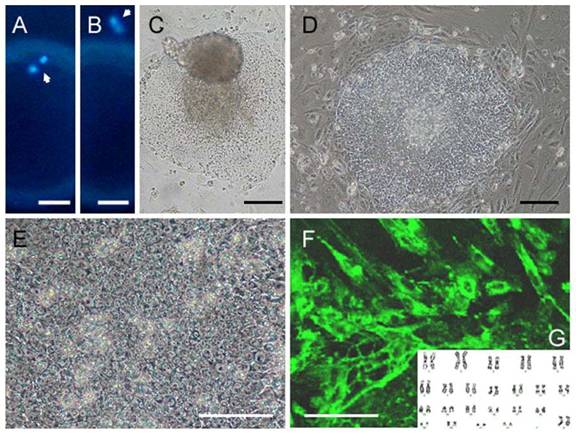
Fig. 2. Expression of characteristic cell surface markers in human SCNT ES cells. SCNT-hES-1 cells expressed cell surface markers including alkaline phosphatase (B), SSEA-3 (H), SSEA-4 (K), TRA-1-60 (N), TRA-1-81 (Q), and Oct-4 (T), but not SSEA-1 (E). The differentiated SCNT-hES-1 cells were not stained with alkaline phosphatase (A). The IVF-derived human ES cells (Miz-hES) were used for comparison and also expressed alkaline phosphatase (C), SSEA-3 (I), SSEA-4 (L), TRA-1-60 (O), TRA-1-81 (R), and Oct-4 (U), but not SSEA-1 (F). Negative controls not treated with first antibodies are shown (D, G, J, M, P, and S). Magnification [(A) to (U), ×40]. Scale bars = 100 µm.
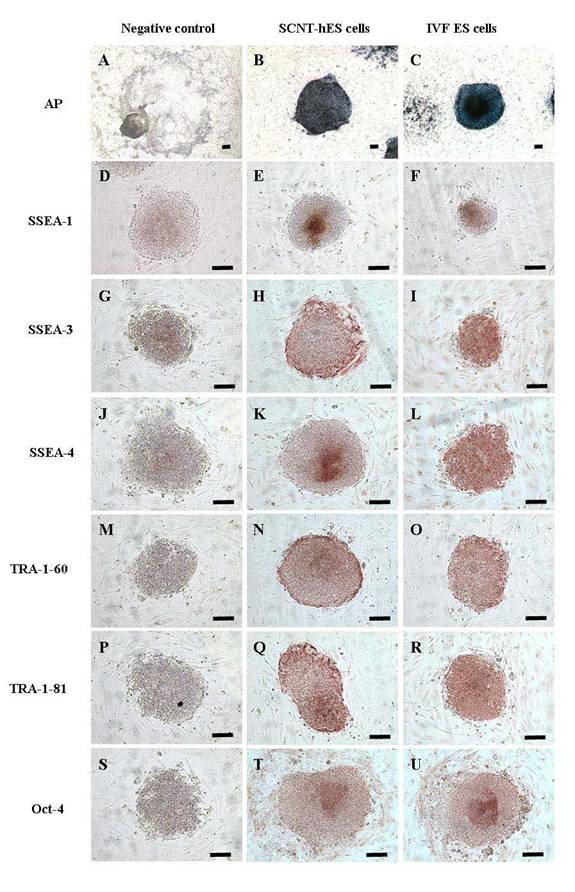
Fig. 3. Teratomas formed by human SCNT ES cells in the testis of SCID mice at twelve weeks after injection. Neuroepithelial rosset (A), pigmented retinal epithelium (B), ostoid island showing bony differentiation (C), cartilage (D), glandular epithelium with smooth muscle and connective tissues (E). Magnification [(A) to (D), ×200; (E), ×100]. Scale bar = 100 µm.


|
|
|