|
Virgin and Child, Enthroned, The Master of Moulins, 1499 |
NUCLEAR
TRANSPLANTATION,
EMBRYONIC
STEM
CELLS,
|
|
|
Virgin and Child, Enthroned, The Master of Moulins, 1499 |
NUCLEAR
TRANSPLANTATION,
EMBRYONIC
STEM
CELLS,
|
|
The New England
Journal of Medicine.
Volume 349:275-286, , July 17, 2003, , Number 3
Nuclear cloning, also referred to as nuclear transfer or nuclear transplantation, denotes the introduction of a nucleus from an adult donor cell into an enucleated oocyte to generate a cloned embryo.[:]
[1] When transferred to the uterus of a female recipient, this embryo has the potential to grow into an infant that is a clone of the adult donor cell, a process termed “reproductive cloning.”
[2] However, when explanted in culture, this embryo can give rise to embryonic stem cells that have the potential to become any or almost any type of cell present in the adult body.
Because embryonic stem cells derived by nuclear transfer are genetically identical to the donor and thus potentially useful for therapeutic applications, this process is called “nuclear transplantation therapy” or “therapeutic cloning.” Therapeutic cloning might substantially improve the treatment of neurodegenerative diseases, blood disorders, or diabetes, since therapy for these diseases is currently limited by the availability or immunocompatibility of tissue transplants. Indeed, experiments in animals have shown that nuclear cloning combined with gene and cell therapy represents a valid strategy for treating genetic disorders.
Reproductive cloning is an inefficient and error-prone process that results in the failure of most clones during development. For a donor nucleus to support development, it must properly activate genes important for early embryonic development and suppress differentiation-associated genes that were transcribed in the original donor cell. Inadequate “reprogramming” of the donor nucleus is thought to be the principal reason for the developmental loss of most clones. In contrast, reprogramming errors do not appear to interfere with therapeutic cloning, because the process appears to select for functional cells.
Recent advances in the field of nuclear cloning allow four major conclusions to be drawn. First, most clones die early in gestation, and only a few survive to birth or beyond. Second, cloned animals have common abnormalities regardless of the type of donor cell or the species used, and third, these abnormalities correlate with aberrant gene expression, which most likely results from faulty genomic reprogramming. Fourth, the efficiency of cloning depends on the state of differentiation of the donor cell. In this article, we will summarize recent results from our laboratory and those of others and review potential therapeutic applications of the nuclear-cloning technology.
THE STATE OF THE ART OF NUCLEAR CLONING
COMMON ABNORMALITIES IN CLONED ANIMALS
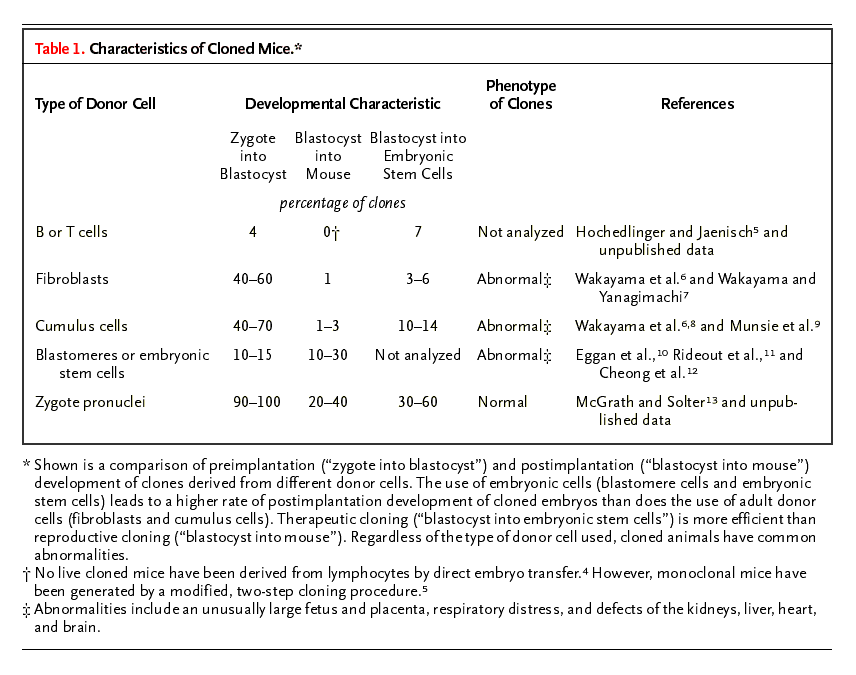
Most cloned embryos die soon after implantation.1,2,3 Those that live to birth often have common abnormalities irrespective of the type of donor cell used (Table 1). For instance, newborn clones are frequently unusually large and have an enlarged placenta (the large-offspring syndrome).2,7,10,14,15,16,17 Moreover, neonate clones often have[:]
respiratory distress
and defects of the kidneys,
liver,
heart,
and brain.18
Even long-term survivors can have abnormalities later in life.
Aging cloned mice were recently reported to[:]
become obese,19
die prematurely, and
have tumors.20
|
|
Table 1. Characteristics of Cloned Mice.
Some of these phenotypes do, however, appear to be specific to the type of donor cell used. For example,[:]
clones derived from cumulus cells (somatic cells that surround the egg) become obese,19
whereas clones derived from Sertoli cells (somatic cells that nourish the egg) die prematurely.20
However, these abnormalities are not inherited by the offspring of the clones, suggesting that epigenetic rather than genetic aberrations are the cause; epigenetic changes, in contrast to genetic changes, are reversible modifications of DNA or chromatin that are usually erased in the germ line. These results indicate that most problems associated with cloning appear to be due to faulty epigenetic reprogramming of the transplanted nucleus.
FAULTY EPIGENETIC REPROGRAMMING in CLONES
Faulty epigenetic reprogramming is the failure to return the gene-expression program of a somatic donor nucleus to an embryonic pattern of expression.2 At the molecular level, epigenetic modifications that are specific to the differentiated cell, such as DNA methylation, histone modifications, and the overall chromatin structure, need to be reprogrammed to a state compatible with embryonic development. Consistent with this notion is the finding that embryos cloned from somatic cells frequently fail to reactivate key embryonic genes at the blastocyst stage.21,22 Moreover, cloned embryos can have aberrant patterns of DNA methylation23,24,25 and precocious expression of genes specific to the donor cell.26 In contrast, embryos cloned from embryonic stem cells faithfully express early embryonic genes,21 possibly because these genes are already active in the donor genome. This might explain why cloning from embryonic stem cells is roughly 10 to 20 times as efficient as cloning from somatic cells (Table 1).
During normal development, reprogramming occurs before and after the formation of the zygote2,27 (Table 2). Faithful reprogramming ensures the proper activation of genes during embryonic development. Prezygotic reprogramming includes the acquisition of genomic imprints — the expression of genes from either the paternal or maternal set of chromosomes — as well as the modification of most somatic genes during gametogenesis. Inactivation of the X chromosome and adjustment of the length of telomeres are examples of postzygotic reprogramming.
|
|
Table 2. Outcome of Epigenetic Reprogramming in Cloned Animals.
PREZYGOTIC REPROGRAMMING
Because cloning uses an unfertilized, mature oocyte, reprogramming has to occur within the brief interval between the transfer of the donor nucleus into the oocyte and the start of zygotic transcription. Thus, prezygotic modifications (i.e., any modifications that have occurred before the mature oocyte stage) are expected to be less efficiently reprogrammed than postzygotic modifications. Consistent with this hypothesis is the fact that aberrant imprints in donor nuclei are usually not corrected in the clones. Genomic imprinting is an epigenetic modification of DNA resulting in the monoallelic and parent-of-origin–specific expression of certain genes. The dysregulation of imprinted genes is particularly pronounced in cloned mice derived from embryonic stem cells, because cultured embryonic stem cells are epigenetically very unstable and frequently gain or lose genomic imprints.16 Cloned mice derived from uncultured cumulus cells with normal imprints also have aberrant expression of imprinted genes,28 suggesting that the dysregulation of imprinted genes is influenced by both the epigenetic state of the donor cell and the nuclear-transfer procedure. Since imprinted genes are important for fetal growth and placental function, aberrant expression of these genes might account for the severely abnormal fetal and placental phenotypes in many clones.
To assess the degree of dysregulation of nonimprinted genes, gene-expression analyses have been performed on newborn clones. These analyses revealed that hundreds of genes are aberrantly expressed in the placentas and livers of cloned mice derived from either cumulus or embryonic stem cells28 (Table 2). Interestingly, a subgroup of these genes was found to be misexpressed exclusively in clones derived from cumulus cells, and another subgroup was aberrantly expressed only in clones derived from embryonic stem cells — a result consistent with the finding that clones derived from different types of donor cells can have different abnormalities.3,19,20 Therefore, prezygotic reprogramming, which affects the expression of imprinted and most nonimprinted genes, appears to be faulty in clones.
POSTZYGOTIC REPROGRAMMING
Telomeres are structures that protect the ends of chromosomes. Telomeres progressively shorten with each cell division, and this shortening has been correlated with cellular and organismal aging. In most cloned animals, the lengths of telomeres are normal or even longer than normal,30,31,32,33 suggesting that cloned embryos faithfully restore telomere length to that of normal embryos (Table 2).
Inactivation of one X chromosome in female cells is a mechanism that ensures equal dosage of X-linked genes in the two sexes. It is accomplished by the random and stable silencing of one of the two X chromosomes early in embryogenesis. In embryos cloned from female somatic cells, the inactive X was found to be reactivated properly, resulting in random X inactivation in the mouse29 (Table 2). Thus, most postzygotic modifications appear to be properly reprogrammed in clones and are therefore not expected to impede the development of clones.
In summary, all available evidence indicates that reproductive cloning, in contrast to normal development or in vitro fertilization, is limited by the fundamental biologic problem of epigenetic reprogramming of the donor nucleus. Specifically, prezygotic modifications that usually occur during gametogenesis are not corrected in the clones. This incomplete reprogramming may result in abnormal phenotypes, aberrant gene expression, and the death of most clones. Consequently, even the rare surviving clones are likely to have at least subtle abnormalities.
DIFFERENTIATION and CLONING EFFICIENCY
The efficiency of obtaining cloned animals from adult donor cells is low in most species. In general, 1 to 3 percent of cloned blastocysts develop completely1,2,7,8 (Table 1). This rate is slightly higher in cows, in which up to 10 percent of cloned embryos develop to term.34 In contrast to the results of cloning involving somatic donor cells, the results of cloning involving embryonic cells such as blastomeres (cells of the cleavage embryo) or embryonic stem cells are more efficient (10 to 30 percent of cloned blastocysts develop successfully)3,10,11,12 (Table 1), suggesting that the state of differentiation of the donor cell directly affects the efficiency of cloning. This observation is consistent with the idea that embryonic cells require less reprogramming of their genome, because the genes essential for early embryonic development are already active. In fact, the transfer of the nucleus of an embryonic cell, such as an embryonic stem cell, might have nearly the same rate of success in generating live-born mice as does transfer of the zygotic nucleus (or pronucleus)13 (Table 1). However, mice cloned from the nuclei of embryonic stem cells, in contrast to mice derived from zygotic nuclei, are abnormal,10 indicating that gametogenesis and fertilization endow zygotic nuclei with the ability to direct normal development. In summary, these data indicate that cells progressively lose nuclear potency as they develop.
TERMINALLY DIFFERENTIATED CELLS REMAIN TOTIPOTENT
The loss of nuclear potency in differentiating cells raised the important question of whether the nuclei of terminally differentiated donor cells retain developmental totipotency — that is, the potential to give rise to an entire organism. Previous nuclear-transfer experiments in frogs35 and mammals36 failed to resolve this question, because of the lack of appropriate genetic markers that would unambiguously identify terminally differentiated cells within a heterogeneous population of donor cells. It is possible that the clones were not derived from differentiated donor cells but rather from adult stem cells that were present in the adult donor animal at low frequencies.3,37,38,39
To address this question, we created monoclonal mice from terminally differentiated lymphocytes.5 The characteristic genetic rearrangements at the immune-receptor loci of mature lymphoid cells served as genetic markers, which allowed us to draw the retrospective and unambiguous conclusion that the clones had been derived from a terminally differentiated donor nucleus. We found that nuclei from mature B and T cells were able to direct development after being transferred into an oocyte, but this process was much less efficient than cloning involving other adult donor cells, such as fibroblasts or cumulus cells6 7,8 (Table 1). Previous attempts to generate monoclonal mice from lymphoid donor nuclei by the direct transfer of blastocysts into the uterus were unsuccessful.4 To derive monoclonal mice from the nuclei of mature B and T cells, we used a two-step cloning procedure in which the derivation of embryonic stem cells from cloned blastocysts was followed by tetraploid embryo complementation.10 In this approach, diploid embryonic stem cells are injected into tetraploid host blastocysts to generate mice. Because tetraploid cells can form a functional placenta but not an embryo, the resultant mice had to have been derived entirely from the injected embryonic stem cells.
Although the generation of monoclonal mice demonstrated unequivocally that terminally differentiated cells can remain genetically totipotent, these results did not exclude the possibility that many cloned animals are derived from less well differentiated adult cells, such as adult stem cells. The genome of adult stem cells might resemble that of embryonic stem cells, which is more amenable to or requires less reprogramming than the genome of a differentiated cell. It will be interesting to test whether purified adult stem cells can serve as efficient somatic donor cells. This question is also of importance with respect to the potential therapeutic application of nuclear transfer; because nuclear transfer is inherently inefficient, it will be essential to identify the most efficient donor cell in the adult in order to reduce the number of oocytes that are needed to establish a line of embryonic stem cells.
THERAPEUTIC POTENTIAL of NUCLEAR TRANSPLANTATION
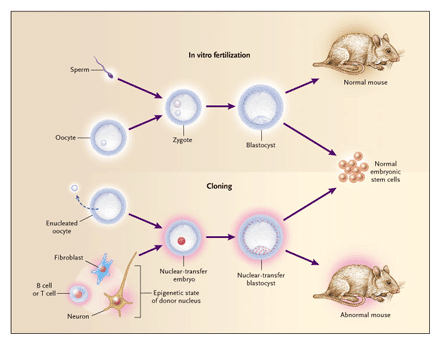
REPRODUCTIVE CLONING VERSUS THERAPEUTIC CLONING
In addition to its value in the study of nuclear changes during differentiation, nuclear-transfer technology has substantial therapeutic potential. For the following discussion, it is important to distinguish between reproductive cloning and nuclear-transplantation therapy (also referred to as therapeutic cloning)40,41 (Figure 1). The purpose of reproductive cloning is to generate a cloned embryo, which is then implanted in the uterus of a female to give rise to a cloned individual. In contrast, the purpose of nuclear-transplantation therapy is to generate an autologous embryonic stem-cell line that is derived from a cloned embryo — referred to as nuclear-transfer embryonic stem cells — and that can be used for tissue replacement.
|
|
Figure 1. Comparison of Normal Development with Development during Reproductive Cloning and Therapeutic Cloning.
During normal development (Panel A), a haploid (1n) sperm cell fertilizes a haploid oocyte to form a diploid (2n) zygote that undergoes cleavage to become a blastocyst embryo. Blastocysts are implanted in the uterus and ultimately give rise to an animal. During reproductive cloning (Panel B), the diploid nucleus of an adult donor cell is introduced into an enucleated oocyte, which after artificial activation divides into a cloned blastocyst. On transfer into surrogate mothers, a few of the cloned blastocysts will give rise to a clone. In contrast, therapeutic cloning (Panel C) requires the explantation of cloned blastocysts in culture to yield a line of embryonic stem cells that can potentially differentiate in vitro into any type of cell for therapeutic purposes.
Rejection is a frequent complication of allogeneic organ transplantation, owing to immunologic incompatibility. To prevent this host-versus-graft disease, immunosuppressive drugs are routinely given to transplant recipients — a treatment that has serious side effects. Embryonic stem cells derived from nuclear transplantation are genetically identical to the patient’s cells, thus eliminating the risk of immune rejection and the requirement for immunosuppression. Moreover, embryonic stem cells provide a renewable source of replacement tissue, allowing therapy to be repeated whenever needed.
DIFFERENTIATION INTO FUNCTIONAL CELLS
Therapeutic cloning requires the in vitro differentiation of nuclear-transfer embryonic stem cells into a homogeneous population of functional cells that can be used for cell therapy. In some circumstances, these cells may first need to be manipulated to correct defects. Recently, protocols have been developed that allow homologous recombination and thus genetic manipulation of human embryonic stem cells.42 Various studies have described the potential of human embryonic stem cells to differentiate into multiple lineages,43,44 such as neural progenitors,45,46,47 hematopoietic precursors,48 and insulin-secreting cells.49
Protocols for the differentiation of murine embryonic stem cells into functional cells of many if not all organs present in adult mice are well established (Table 3). For example, embryonic stem cells can generate functional motor neurons when they are exposed to signals that normally induce neurogenesis.50 With a different strategy, drug-selection protocols have been used to cause embryonic stem cells to differentiate into homogeneous populations of cardiomyocytes,54 neuroepithelial precursor cells,55 and insulin-producing cells.52 The expression of the homeobox protein HoxB4 in embryoid bodies generates hematopoietic stem cells that avert death in mice that have received lethal doses of radiation.53 Similarly, the expression of nuclear receptor–related factor 1 (Nurr1), an orphan nuclear receptor that is expressed chiefly in the central nervous system, in embryonic stem cells induces the formation of dopaminergic neurons that can relieve behavioral symptoms in rats with Parkinson’s disease.51
|
|
COMBINING NUCLEAR CLONING WITH GENE AND CELL THERAPY
The ultimate goal of therapeutic cloning is to generate functional cells from cloned embryonic stem cells that can be used for cell transplantation in patients. Several groups, including ours, have shown that nuclear-transfer embryonic stem cells can be derived from mouse cumulus or fibroblast cells and that these cells can be coaxed into becoming somatic cells such as myogenic cells, dopaminergic and serotonergic neurons, or hematopoietic cells.5,6,9,56 However, before these principles can be applied clinically, it is important to demonstrate the feasibility of therapeutic cloning in an animal model of disease.
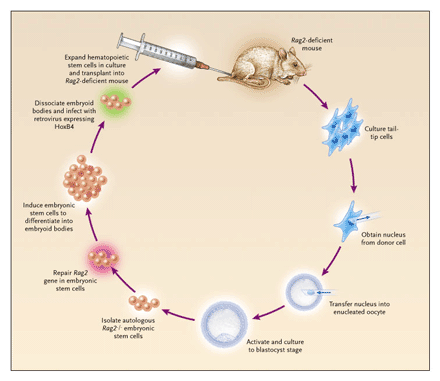
In an attempt to establish such a mouse model, we have combined nuclear cloning with gene and cell therapy to treat a genetic disorder (Figure 2).56 We chose the well-characterized Rag2 mutant mouse, which has severe combined immunodeficiency owing to a mutation in the recombination-activating gene 2 (Rag2), which catalyzes immune-receptor rearrangements in lymphocytes. This mouse is devoid of mature B and T cells, a condition resembling Omenn’s syndrome in humans. First, we isolated somatic (fibroblast) cells from the tails of Rag2-deficient mice and injected the nuclei of these cells into enucleated eggs. We then cultured the resultant embryos to the blastocyst stage and isolated the autologous embryonic stem cells. Subsequently, one of the mutant Rag2 alleles was targeted by homologous recombination in embryonic stem cells in order to restore normal gene structure. To obtain somatic cells for treatment, these embryonic stem cells underwent differentiation into embryoid bodies (embryo-like structures that contain various types of somatic cells) and further into hematopoietic precursors by expressing HoxB4. The resulting hematopoietic precursors were transplanted into irradiated Rag2-deficient animals to treat the disease.
|
|
Figure 2. Mouse Model of Therapeutic Cloning.
Tail-tip cells were obtained from mice with a deficiency of recombination-activating gene 2 (Rag2) and cultured, and the nuclei were transferred into enucleated oocytes. The cloned embryos were cultured to the blastocyst stage to derive autologous embryonic stem cells. After one of the mutant Rag2 alleles was repaired by homologous recombination, embryonic stem cells were induced to differentiate into embryoid bodies (embryo-like structures that contain various types of somatic cells) and infected with a retrovirus expressing the homeobox protein HoxB4. The resultant hematopoietic stem cells were clonally expanded and injected intravenously into irradiated Rag2-deficient animals to reconstitute their immune system. Adapted from Rideout et al.,56 with the permission of the publisher.
The initial attempts to engraft these cells were unsuccessful because of an increased level of natural killer cells in the mutant host. Hematopoietic cells derived from embryonic stem cells express low levels of major-histocompatibility-complex class I molecules and are thus a preferred target of destruction by natural killer cells. Elimination of the natural killer cells by antibody depletion or genetic ablation allowed the nuclear-transfer embryonic stem cells to differentiate into the myeloid lineages efficiently and, to a lesser degree, into the lymphoid lineages. Functional B and T cells whose immunoglobulin and T-cell–receptor alleles had been properly rearranged were detected in the mice, as were serum immunoglobulins. However, because HoxB4 appears to promote the differentiation of embryonic stem cells into myeloid cells, lymphoid reconstitution might be more successful if transcription factors specific to the lymphoid lineage were used.
This experiment demonstrated that nuclear transfer can be combined with gene therapy to treat a genetic disorder. Consequently, therapeutic cloning should be useful in other diseases in which the genetic cause is known, such as sickle cell anemia and -thalassemia.
LIMITATIONS and ALTERNATIVES
FAULTY REPROGRAMMING in CLONES as a POTENTIAL IMPEDIMENT to THERAPEUTIC APPLICATIONS
An important question is whether the reprogramming errors leading to abnormal phenotypes in cloned animals would impede the therapeutic use of nuclear-transfer technology. For the derivation of embryonic stem cells from fertilized embryos, blastocysts are explanted in vitro and cultured until a small colony forms that can be dissociated. Only one or a few of the dissociated cells have the potential to grow into an embryonic stem-cell line,57 suggesting that competent cells are selected for in culture. Similarly, the derivation of embryonic stem cells from cloned blastocysts may be the result of selection for a few successfully reprogrammed cells within a cloned embryo (Figure 3). In contrast, the development of a cloned embryo after implantation most likely does not allow for the in vivo selection of a few functional cells, thus causing developmental failure of the clone or phenotypic abnormalities. In support of this notion, the derivation of embryonic stem cells from somatic donor cells is more efficient5 6,9,56 than is the generation of cloned mice4 7,8 (Table 1).
|
|
Figure 3. Derivation of Embryonic Stem Cells from a Blastocyst, Resulting in Selection for Functional Cells.
Only a few of the cells of a blastocyst derived from a fertilized zygote have the potential to produce an embryonic stem-cell line, and these cells seem to be selected for by culture conditions (top panel). Mice derived from fertilized zygotes are normal because the sperm and oocyte genomes have undergone proper reprogramming during gametogenesis (top panel). However, cloned blastocysts and the resultant mice seem to maintain a memory of the epigenetic state of the donor nucleus they were derived from (bottom panel). This is probably due to faulty reprogramming of the somatic donor nucleus after nuclear transfer and results in abnormal phenotypes and aberrant patterns of gene expression. These epigenetic abnormalities are represented by the pink halo surrounding the cloned embryo and mouse. In contrast, the derivation of embryonic stem cells from cloned blastocysts appears to select for fully reprogrammed, functional cells that have lost this epigenetic memory.
Abnormal fetal development is the most fundamental cause of clone failure. In contrast to the result of reproductive cloning, no fetus is formed in therapeutic cloning. Thus, aberrant expression of genes that are essential for normal fetal development, such as imprinted genes, is not expected to impede the functionality of embryonic stem cells that undergo differentiation in vitro. The abnormal expression of some imprinted genes, such as the gene for insulin-like growth factor 2, however, has been associated with disease in the adult,58,59 and it will be important to determine whether dysregulation of these genes has adverse effects on the function of somatic cells derived from embryonic stem cells.
Nuclear-transfer embryonic stem cells have the same developmental potency as embryonic stem cells derived from fertilized eggs. When injected into blastocysts, nuclear-transfer embryonic stem cells contributed to tissues of all three germ layers, including the germ line.6 A subgroup of these embryonic stem-cell lines was even able to produce mice after tetraploid embryo complementation,5,56 a process that allows the generation of mice from embryonic stem cells alone. It is notable that the abnormalities regularly associated with cloned animals were not observed in these mice.
In any therapeutic setting, cells derived from nuclear-transfer embryonic stem cells will be introduced into a patient with a disease, and host cells will interact with the transplanted cloned cells to generate a chimeric tissue. Chimeric animals generated by the injection of normal or nuclear-transfer embryonic stem cells into normal blastocysts form normal chimeras.6,9 This finding suggests that the presence of host helper cells, which are derived from the fertilized egg, complement the defects that invariably result when a cloned animal is generated from a somatic or embryonic donor nucleus. In support of this notion, Byrne et al. showed that embryonic cells derived from nuclear transfer failed to develop on their own in frogs but integrated normally when they were combined with wild-type embryos to form chimeric tadpoles.60 In summary, these considerations suggest that nuclear-transfer embryonic stem cells are equivalent to embryonic stem cells derived from a fertilized zygote.
ADULT STEM CELLS as an ALTERNATIVE to THERAPEUTIC CLONING
Are there alternatives to therapeutic cloning? Adult stem cells are another potential source of autologous cells for transplantation therapy. They have been isolated from adult tissues such as brain, bone marrow, skin, and muscle, and they might have a broader developmental potential than originally anticipated.61 However, it remains unclear whether the observed plasticity, or “transdifferentiation potential,” of adult stem cells is inherent to the cells or the consequence of culture conditions, contamination, or cell fusion.61,62,63,64,65 Moreover, recent experiments have failed to reproduce the results of some earlier reports claiming that transdifferentiation occurred.66,67
The therapeutic potential of adult stem cells appears to be much lower than that of embryonic stem cells. First, adult stem cells are difficult to isolate and hard to propagate in culture. In contrast, embryonic stem cells are derived rather easily (once an embryo has been obtained), and they grow indefinitely in culture. Second, embryonic stem cells can be manipulated genetically by homologous recombination to correct a genetic defect.56 In contrast, currently, adult stem cells can be genetically manipulated only through the introduction of retroviral transgenes, which overexpress genes at variable levels and can lead to insertional mutagenesis and cancer.68 Third, embryonic stem cells can be coaxed into becoming any type of cell through the use of specific culture conditions or genetic manipulation. The differentiation potential of adult stem cells, however, seems to be restricted.
One notable exception in this respect is the recent isolation of multipotent adult progenitor cells.69 Multipotent adult progenitor cells were derived from the bone marrow of adult mice, rats, and humans after a three-month culture protocol. These cells have the potential to differentiate into cells of all three germ layers both in vitro and in vivo after being injected into blastocysts. However, it has not been demonstrated in animal models or humans that multipotent adult progenitor cells can be used to correct a disease phenotype.
THE REQUIREMENT for HUMAN OOCYTES
To overcome the ethical and practical limitations of therapeutic cloning, it would be useful to reprogram somatic cells directly into embryonic stem cells without the use of oocytes. An understanding of the factors that have a role in establishing and maintaining pluripotency might make it possible to alter the fate of somatic cells directly. For instance, the embryonic transcription factor Oct-4 appears to act as a regulator of pluripotency during development.57,70 When mutated, embryos cannot form a pluripotent inner cell mass, and their development is arrested.71 Thus, manipulation of Oct-4 and related genes21 in somatic cells might help to reprogram their nuclei to an embryonic state. This could reduce or even circumvent the need for human oocytes.
Recently, Hübner et al. provided evidence of the differentiation of murine embryonic stem cells into oocyte-like cells in vitro.72 It will be interesting to determine whether oocytes can be obtained from human embryonic stem cells and whether these cells are suitable for nuclear transfer.
CONCLUSIONS
Therapeutic cloning, in combination with the differentiation potential of embryonic stem cells, offers a valuable means of obtaining autologous cells for the treatment of a variety of diseases. The abnormalities associated with reproductive cloning are not expected to impede the use of this technique for therapy, since the process seems to select for functional cells. However, before these principles can be applied clinically, it will be essential to improve differentiation protocols for human embryonic stem cells and to evaluate the effect of oocyte-derived mitochondrial proteins in somatic cells obtained by nuclear transfer. In the future it might be possible to generate embryonic stem cells directly from somatic cells. It is important, therefore, to continue research aimed at improving our understanding of the molecular events that take place during nuclear reprogramming, in order to develop these potential new therapies.
Supported by a Ph.D. fellowship from the Boehringer Ingelheim Fonds (to Dr. Hochedlinger) and by a grant (R37-CA84198, to Dr. Jaenisch) from the National Cancer Institute.
We are indebted to Caroline Beard, Robert Blelloch, Kevin Eggan, Joost Gribnau, and Teresa Holm for discussions and critical reading of the manuscript.
Source Information
From the Whitehead Institute for Biomedical Research (K.H., R.J.) and the Department of Biology, Massachusetts Institute of Technology (R.J.) — both in Cambridge.
Address reprint requests to Dr. Jaenisch at the Whitehead Institute, 9 Cambridge Center, Cambridge, MA 02142, or at jaenisch@wi.mit.edu.
References
REFERENCES
1. Solter D. Mammalian cloning: advances and limitations. Nat Rev Genet 2000;1:199-207.[CrossRef][ISI][Medline]
2. Rideout WM III, Eggan K, Jaenisch R. Nuclear cloning and epigenetic reprogramming of the genome. Science 2001;293:1093-1098.[Abstract/Full Text]
3. Hochedlinger K, Jaenisch R. Nuclear transplantation: lessons from frogs and mice. Curr Opin Cell Biol 2002;14:741-748.[CrossRef][ISI][Medline]
4. Wakayama T, Yanagimachi R. Mouse cloning with nucleus donor cells of different age and type. Mol Reprod Dev 2001;58:376-383.[CrossRef][ISI][Medline]
5. Hochedlinger K, Jaenisch R. Monoclonal mice generated by nuclear transfer from mature B and T donor cells. Nature 2002;415:1035-1038.[CrossRef][ISI][Medline]
6. Wakayama T, Tabar V, Rodriguez I, Perry AC, Studer L, Mombaerts P. Differentiation of embryonic stem cell lines generated from adult somatic cells by nuclear transfer. Science 2001;292:740-743.[Abstract/Full Text]
7. Wakayama T, Yanagimachi R. Cloning of male mice from adult tail-tip cells. Nat Genet 1999;22:127-128.[CrossRef][ISI][Medline]
8. Wakayama T, Perry AC, Zuccotti M, Johnson KR, Yanagimachi R. Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature 1998;394:369-374.[CrossRef][ISI][Medline]
9. Munsie MJ, Michalska AE, O’Brien CM, Trounson AO, Pera MF, Mountford PS. Isolation of pluripotent embryonic stem cells from reprogrammed adult mouse somatic cell nuclei. Curr Biol 2000;10:989-992.[CrossRef][ISI][Medline]
10. Eggan K, Akutsu H, Loring J, et al. Hybrid vigor, fetal overgrowth, and viability of mice derived by nuclear cloning and tetraploid embryo complementation. Proc Natl Acad Sci U S A 2001;98:6209-6214.[Abstract/Full Text]
11. Rideout WM III, Wakayama T, Wutz A, et al. Generation of mice from wild-type and targeted ES cells by nuclear cloning. Nat Genet 2000;24:109-110.[CrossRef][ISI][Medline]
12. Cheong HT, Takahashi Y, Kanagawa H. Birth of mice after transplantation of early cell-cycle-stage embryonic nuclei into enucleated oocytes. Biol Reprod 1993;48:958-963.[Abstract]
13. McGrath J, Solter D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 1984;37:179-183.[ISI][Medline]
14. Tanaka S, Oda M, Toyoshima Y, et al. Placentomegaly in cloned mouse concepti caused by expansion of the spongiotrophoblast layer. Biol Reprod 2001;65:1813-1821.[Abstract/Full Text]
15. Hill JR, Burghardt RC, Jones K, et al. Evidence for placental abnormality as the major cause of mortality in first-trimester somatic cell cloned bovine fetuses. Biol Reprod 2000;63:1787-1794.[Abstract/Full Text]
16. Young LE, Sinclair KD, Wilmut I. Large offspring syndrome in cattle and sheep. Rev Reprod 1998;3:155-163.[ISI][Medline]
17. Humpherys D, Eggan K, Akutsu H, et al. Epigenetic instability in ES cells and cloned mice. Science 2001;293:95-97.[Abstract/Full Text]
18. Cibelli JB, Campbell KH, Seidel GE, West MD, Lanza RP. The health profile of cloned animals. Nat Biotechnol 2002;20:13-14.[CrossRef][ISI][Medline]
19. Tamashiro KL, Wakayama T, Akutsu H, et al. Cloned mice have an obese phenotype not transmitted to their offspring. Nat Med 2002;8:262-267.[CrossRef][ISI][Medline]
20. Ogonuki N, Inoue K, Yamamoto Y, et al. Early death of mice cloned from somatic cells. Nat Genet 2002;30:253-254.[CrossRef][ISI][Medline]
21. Bortvin A, Eggan K, Skaletsky H, et al. Incomplete reactivation of Oct4-related genes in mouse embryos cloned from somatic nuclei. Development 2003;130:1673-1680.[Abstract/Full Text]
22. Boiani M, Eckardt S, Scholer HR, McLaughlin KJ. Oct4 distribution and level in mouse clones: consequences for pluripotency. Genes Dev 2002;16:1209-1219.[Abstract/Full Text]
23. Kang YK, Koo DB, Park JS, et al. Aberrant methylation of donor genome in cloned bovine embryos. Nat Genet 2001;28:173-177.[CrossRef][ISI][Medline]
24. Kang YK, Park JS, Koo DB, et al. Limited demethylation leaves mosaic-type methylation states in cloned bovine pre-implantation embryos. EMBO J 2002;21:1092-1100.[Abstract/Full Text]
25. Dean W, Santos F, Stojkovic M, et al. Conservation of methylation reprogramming in mammalian development: aberrant reprogramming in cloned embryos. Proc Natl Acad Sci U S A 2001;98:13734-13738.[Abstract/Full Text]
26. Gao S, Chung YG, Williams JW, Riley J, Moley K, Latham KE. Somatic cell-like features of cloned mouse embryos prepared with cultured myoblast nuclei. Biol Reprod (in press).
27. Jaenisch R, Eggan K, Humpherys D, Rideout W, Hochedlinger K. Nuclear cloning, stem cells, and genomic reprogramming. Cloning Stem Cells 2002;4:389-96.
28. Humpherys D, Eggan K, Akutsu H, et al. Abnormal gene expression in cloned mice derived from embryonic stem cell and cumulus cell nuclei. Proc Natl Acad Sci U S A 2002;99:12889-12894.[Abstract/Full Text]
29. Eggan K, Akutsu H, Hochedlinger K, Rideout W III, Yanagimachi R, Jaenisch R. X-chromosome inactivation in cloned mouse embryos. Science 2000;290:1578-1581.[Abstract/Full Text]
30. Betts D, Bordignon V, Hill J, et al. Reprogramming of telomerase activity and rebuilding of telomere length in cloned cattle. Proc Natl Acad Sci U S A 2001;98:1077-1082.[Abstract/Full Text]
31. Lanza RP, Cibelli JB, Blackwell C, et al. Extension of cell life-span and telomere length in animals cloned from senescent somatic cells. Science 2000;288:665-669.[Abstract/Full Text]
32. Tian XC, Xu J, Yang X. Normal telomere lengths found in cloned cattle. Nat Genet 2000;26:272-273.[CrossRef][ISI][Medline]
33. Wakayama T, Shinkai Y, Tamashiro KL, et al. Cloning of mice to six generations. Nature 2000;407:318-319.[CrossRef]
34. Wells DN, Misica PM, Tervit HR. Production of cloned calves following nuclear transfer with cultured adult mural granulosa cells. Biol Reprod 1999;60:996-1005.[Abstract/Full Text]
35. Gurdon JB, Laskey RA, Reeves OR. The developmental capacity of nuclei transplanted from keratinized skin cells of adult frogs. J Embryol Exp Morphol 1975;34:93-112.[ISI][Medline]
36. Wilmut I, Schnieke AE, McWhir J, Kind AJ, Campbell KH. Viable offspring derived from fetal and adult mammalian cells. Nature 1997;385:810-813. [Erratum, Nature 1997;386:200.][CrossRef][ISI][Medline]
37. Liu L. Cloning efficiency and differentiation. Nat Biotechnol 2001;19:406-406.[CrossRef][ISI][Medline]
38. Oback B, Wells D. Donor cells for nuclear cloning: many are called, but few are chosen. Cloning Stem Cells 2002;4:147-68.
39. Weissman IL. Stem cells: units of development, units of regeneration, and units in evolution. Cell 2000;100:157-168.[ISI][Medline]
40. Vogelstein B, Alberts B, Shine K. Genetics: please don’t call it cloning! Science 2002;295:1237-1237.[Full Text]
41. Colman A, Kind A. Therapeutic cloning: concepts and practicalities. Trends Biotechnol 2000;18:192-196.[CrossRef][ISI][Medline]
42. Zwaka TP, Thomson JA. Homologous recombination in human embryonic stem cells. Nat Biotechnol 2003;21:319-321.[CrossRef][ISI][Medline]
43. Schuldiner M, Yanuka O, Itskovitz-Eldor J, Melton DA, Benvenisty N. Effects of eight growth factors on the differentiation of cells derived from human embryonic stem cells. Proc Natl Acad Sci U S A 2000;97:11307-11312.[Abstract/Full Text]
44. Odorico JS, Kaufman DS, Thomson JA. Multilineage differentiation from human embryonic stem cell lines. Stem Cells 2001;19:193-204.[Abstract/Full Text]
45. Carpenter MK, Inokuma MS, Denham J, Mujtaba T, Chiu CP, Rao MS. Enrichment of neurons and neural precursors from human embryonic stem cells. Exp Neurol 2001;172:383-397.[CrossRef][ISI][Medline]
46. Reubinoff BE, Itsykson P, Turetsky T, et al. Neural progenitors from human embryonic stem cells. Nat Biotechnol 2001;19:1134-1140.[CrossRef][ISI][Medline]
47. Schuldiner M, Eiges R, Eden A, et al. Induced neuronal differentiation of human embryonic stem cells. Brain Res 2001;913:201-205.[CrossRef][ISI][Medline]
48. Kaufman DS, Hanson ET, Lewis RL, Auerbach R, Thomson JA. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proc Natl Acad Sci U S A 2001;98:10716-10721.[Abstract/Full Text]
49. Assady S, Maor G, Amit M, Itskovitz-Eldor J, Skorecki KL, Tzukerman M. Insulin production by human embryonic stem cells. Diabetes 2001;50:1691-1697.[Abstract/Full Text]
50. Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell 2002;110:385-397.[ISI][Medline]
51. Kim JH, Auerbach JM, Rodriguez-Gomez JA, et al. Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson’s disease. Nature 2002;418:50-56.[CrossRef][ISI][Medline]
52. Soria B, Roche E, Berna G, Leon-Quinto T, Reig JA, Martin F. Insulin-secreting cells derived from embryonic stem cells normalize glycemia in streptozotocin-induced diabetic mice. Diabetes 2000;49:157-162.[Abstract]
53. Kyba M, Perlingeiro RC, Daley GQ. HoxB4 confers definitive lymphoid-myeloid engraftment potential on embryonic stem cell and yolk sac hematopoietic progenitors. Cell 2002;109:29-37.[ISI][Medline]
54. Klug MG, Soonpaa MH, Koh GY, Field LJ. Genetically selected cardiomyocytes from differentiating embryonic stem cells form stable intracardiac grafts. J Clin Invest 1996;98:216-224.[Abstract/Full Text]
55. Li M, Pevny L, Lovell-Badge R, Smith A. Generation of purified neural precursors from embryonic stem cells by lineage selection. Curr Biol 1998;8:971-974.[ISI][Medline]
56. Rideout WM III, Hochedlinger K, Kyba M, Daley GQ, Jaenisch R. Correction of a genetic defect by nuclear transplantation and combined cell and gene therapy. Cell 2002;109:17-27.[ISI][Medline]
57. Buehr M, Nichols J, Stenhouse F, et al. Rapid loss of oct-4 and pluripotency in cultured rodent blastocysts and derivative cell lines. Biol Reprod 2003;68:222-229.[Abstract/Full Text]
58. Reik W, Maher ER. Imprinting in clusters: lessons from Beckwith-Wiedemann syndrome. Trends Genet 1997;13:330-334.[CrossRef][ISI][Medline]
59. Moorehead RA, Sanchez OH, Baldwin RM, Khokha R. Transgenic overexpression of IGF-II induces spontaneous lung tumors: a model for human lung adenocarcinoma. Oncogene 2003;22:853-857.[CrossRef][ISI][Medline]
60. Byrne JA, Simonsson S, Gurdon JB. From intestine to muscle: nuclear reprogramming through defective cloned embryos. Proc Natl Acad Sci U S A 2002;99:6059-6063.[Abstract/Full Text]
61. Joshi CV, Enver T. Plasticity revisited. Curr Opin Cell Biol 2002;14:749-755.[CrossRef][ISI][Medline]
62. Ying QL, Nichols J, Evans EP, Smith AG. Changing potency by spontaneous fusion. Nature 2002;416:545-548.[CrossRef][ISI][Medline]
63. Terada N, Hamazaki T, Oka M, et al. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 2002;416:542-545.[CrossRef][ISI][Medline]
64. Vassilopoulos G, Wang P-R, Russell DW. Transplanted bone marrow regenerates liver by cell fusion. Nature 2003;422:901-904.[CrossRef][ISI][Medline]
65. Wang X, Willenbring H, Akkari Y, et al. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature 2003;422:897-901.[CrossRef][ISI][Medline]
66. Morshead CM, Benveniste P, Iscove NN, van der Kooy D. Hematopoietic competence is a rare property of neural stem cells that may depend on genetic and epigenetic alterations. Nat Med 2002;8:268-273.[CrossRef][ISI][Medline]
67. Wagers AJ, Sherwood RI, Christensen JL, Weissman IL. Little evidence for developmental plasticity of adult hematopoietic stem cells. Science 2002;297:2256-2259.[Abstract/Full Text]
68. Check E. Second cancer case halts gene-therapy trials. Nature 2003;421:305-305.[CrossRef]
69. Jiang Y, Jahagirdar BN, Reinhardt RL, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 2002;418:41-49.[CrossRef][ISI][Medline]
70. Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet 2000;24:372-376.[CrossRef][ISI][Medline]
71. Nichols J, Zevnik B, Anastassiadis K, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell 1998;95:379-391.[ISI][Medline]
72. Hübner K, Fuhrmann G, Christenson LK, et al. Derivation of oocytes from mouse embryonic stem cells. Science 2003;300:1251-1256.[Abstract/Full Text]